

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
It has been well established that attenuated mitochondrial oxidative phosphorylation and cellular respiration is associated with age-related diseases such as obesity, diabetes, neurological disorders including Alzheimer's and Parkinson's diseases, and proliferative cancers. The mitochondrial pyruvate dehydrogenase complex (PDC) has been implicated as a potential therapeutic target because of its activity as a key modulator of energy and metabolic homeostasis [1]. Pyruvate, the primary substrate of PDC, can be utilized for the citric acid cycle after glycolysis or be converted into glucose, glycerol, fatty acids, and non-essential amino acids. This is determined by (1) the efficiency of the mitochondrial pyruvate carrier, (2) the inhibitory effect of pyruvate dehydrogenase kinase (PDK) on PDC, or (3) an alternative pathway where the pyruvate carboxylase (PC) activity is correlated with the rate of gluconeogenesis [2].
The concept of “metabolic inflexibility” describes the failure of metabolism in insulin-resistant human subjects who are not able to respond appropriately to nutritional cues, in terms of adjusting fuel oxidation. Hence, the efflux of acetyl-coenzyme A (CoA) from excess β-oxidation via carnitine acetyltransferase restores insulin function by countering the inhibitory effect on PDC [3]. Many preclinical studies have highlighted the beneficial effect of using dichloroacetate (DCA) in combination therapy when tumor resistance has developed to standard platinum-class drugs alone, which mediates the redirection of glucose metabolism from lactate production to complete oxidation followed by the induction of caspase-mediated apoptosis [4]. The multifunctional PDC regulates metabolic reprogramming involved in pyruvate metabolism to maintain homeostasis and survival. Additionally, the existence of nuclear PDC highlights the complexity of mitochondrial PDC regulation beyond cellular respiration [5]. Hence, this review summarizes the recent findings on PDC regulation in differential tissues and the metabolic flux following the modulated PDC activities in diverse metabolic environments.
REVERSIBLE PHOSPHORYLATION BY PDK AND PDP
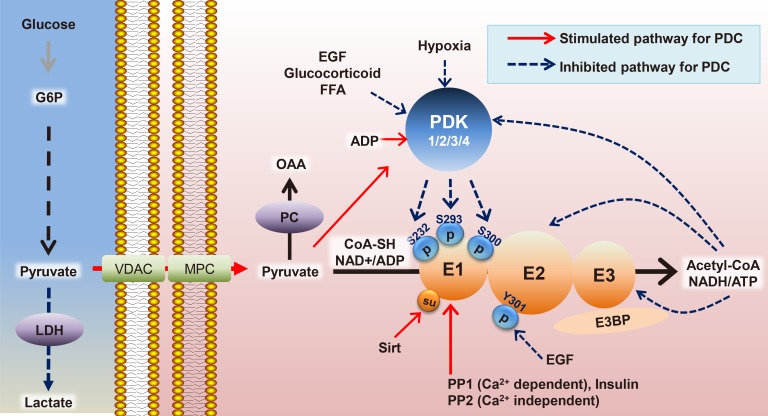
PDK 1-4 which are serine-specific kinases ranging from 45 to 48 kDa are the main players that phosphorylate and inactivate pyruvate dehydrogenase (PDH) E1α. PDK2 exhibits the highest phosphorylation activity on Ser293 of PDH E1α, followed by PDK4, PDK1, and then PDK3 [6]. However, Ser232 is specifically phosphorylated by PDK1 under acidic conditions resulting from the excessive production of acidic metabolites including lactate, protons, and CO2 through increased glycolysis and glutaminolysis [7]. The hypoxia-inducible factor-1α (HIF1α)-mediated PDK1 overexpression reduces the oxidation of glucose and glutamine while increasing the reductive isocitrate dehydrogenase flux, causing reductive carboxylation of glutamine into citrate for proliferation, which suggests metabolic reprogramming [8].
The kinetic activity of PDK is stimulated rapidly by adenosine triphosphate (ATP), nicotinamide adenine dinucleotide (NADH), and acetyl-CoA, and is inhibited by adenosine diphosphate (ADP), NAD+, coenzyme A-SH, and pyruvate in a by-product feedback inhibition manner [7]. The Ca2+-sensitive isoform (pyruvate dehydrogenase phosphatase 1 [PDP1]) is highly expressed in rat heart, brain, and testis, but only the Ca2+-insensitive isoform (PDP2) is regulated by starvation, in the heart [9].
PDK isoforms are increased in metabolic diseases including obesity, diabetes, heart failure, and cancer, and upregulated PDK2 is a potential target for improving glucose tolerance and reducing hepatic steatosis [10]. Many approaches to improve PDC activity have been tested using various PDK inhibitors. However, a deeper understanding of various regulatory factors on PDC activity is still required to design novel targets for numerous metabolic diseases.
TRANSLATIONAL APPROACH THROUGH ACETYLATION AND SUCCINYLATION
Protein posttranslational modifications play an important role in regulating cellular processes. These modifications include acetylation, methylation, biotinylation, ubiquitination, butyrylation, propionylation, crotonylation, malonylation, and succinylation, which can be identified with the help of high-resolution mass spectrometry [11]. Lysine residues in particular are targets for many posttranslational modifications like acetylation and succinylation, which are abundantly identified and studied in different PDC components for their involvement in energy metabolism [12]. According to protein complex analysis of the Lys succinylation proteome, sirtuin 5 (SIRT5) directly and negatively regulates PDH E1α by de-succinylation [11]. A high NADH/NAD+ ratio and acetyl-CoA/CoA ratio causes a reduction in the number of lipoyl groups on the dihydrolipoamide dehydrogenase subunit (E3) and promotes the acetylation of the reduced lipoyl groups by the dihydrolipoamide acetyltransferase subunit (E2) [13].
Mitochondrial acetylation levels are counter-regulated by acetyl-CoA acetyltransferase 1 (ACAT1) and SIRT3 as the upstream acetyltransferase and deacetylase, respectively. The enzymes act on both PDH E1α at K321 and PDP1 at K202, resulting in the recruitment of PDK1 and the dissociation of PDP1 from PDH E1α. They are separately dependent on the level of acetyl-CoA and succinyl-CoA, and both can be reversed by members of the SIRT family [14]. Large-scale quantitative analysis of the global proteome, acetylome, and succinylome using high sensitivity mass spectrometry and bioinformatics demonstrates that both acetylation and succinylation affect specific enzymes of central carbon metabolism including PDH, mediated by the anti-tumoral effect of DCA [15]. Based on the growing data set of acetylation and succinylation in various proteins, modification of residues on PDC should be assessed to show their significance in the overall function.
REGULATORY METABOLITES
The higher levels of reactive molecular species including metabolic intermediates (acetyl-CoA and succinyl-CoA) and reactive oxygen species (ROS) can modulate enzymatic activities involved in the glycolytic pathway and the Randle cycle corresponding to preferential fatty acid oxidation (FAO) [7]. The acetyl-CoA is bound to histone tails by histone acetyltransferase in order to deliver metabolic information on chromatin dynamics as an epigenetic control, and dynamic histone acetylation represents the metabolic status of glucose metabolism, confirmed by energetic heatmap expression patterns of the glycolytic and tricarboxylic acid (TCA) pathways [16]. Likewise, higher levels of acetyl-CoA leading to more malonyl-CoA can inhibit FAO because of a direct feedback loop [17]. Meanwhile, the oxidative stress in type 2 diabetes mellitus (T2DM) reprograms glucose oxidation to lipid oxidation by upregulating the pentose pathway via molecules such as glucose-6-phosphate dehydrogenase and nicotinamide adenine dinucleotide phosphate (NADPH), which can be corrected by apocynin, an antioxidant [18]. The peroxisome proliferator-activated receptor α (PPARα)- or retinoid X receptor (RXR)-induced PDK4 expression is mostly responsive to the increased NADH/NAD+ ratios from mitochondrial β-oxidation [19]. Glucose is utilized not only to generate ATP by glycolysis, but also as a building block for other macromolecule biosynthesis, which would make it a beneficial target for abnormal smooth muscle cells and proliferative cancer cells [20]. Recent application of 13C-radiolabeled metabolomics in patients using high quality technology for the detection of small molecules with high sensitivity will allow us to develop novel therapeutic targets for its modulation in many metabolic disorders. We schematized about PDC regulations generally in Fig. 1.
REGULATION OF PDC IN DIFFERENT ORGANS
Heart
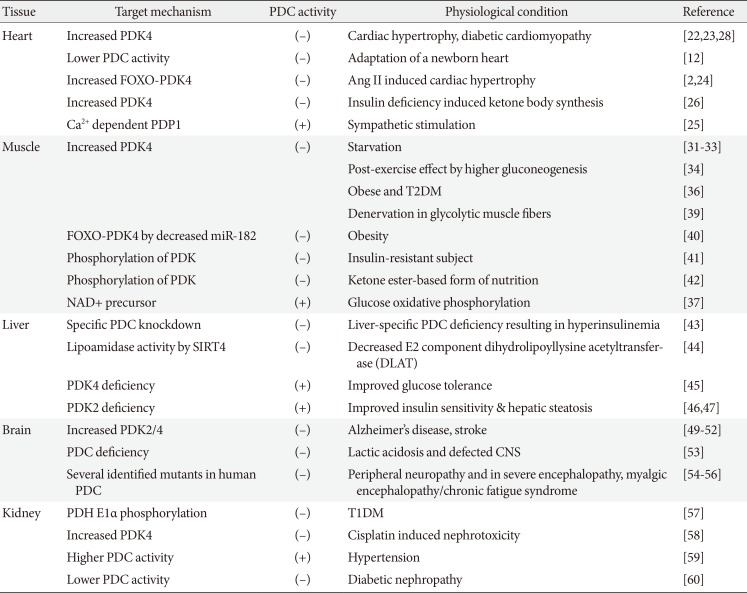
The heart prefers to utilize the oxidation of fatty acids as the primary energy source over glucose for cardiac output (accounting for 60% to 90%), which is a high ATP-consuming process [21]. The limitation of metabolic flexibility to oxidize glucose can contribute to the development of cardiac hypertrophy and diabetic cardiomyopathy because of prolonged reliance on FAO through allosteric inhibitory regulation of the key glycolytic enzymes such as phophofructokinase-2 and PDK4, especially in the current epidemic of obesity and diabetes [22]. Non-enzymatic reactions of acetyl-CoA with PDH using an acetylating agent inhibit pyruvate oxidation derived from hyperacetylation of protein lysine residues in cardiomyopathy models, which is similar to the rodent model of chronic type 1 diabetes mellitus (T1DM), characterized by metabolic inflexibility and decreased contractility [23]. A dramatic metabolic shift from glycolysis to FAO during the adaptation of a newborn heart is mediated by the increased acetylation of glycolytic enzymes, while PDH is inhibited by hyperphosphorylation and hypersuccinylation [12]. The enhanced Forkhead box O (FoxO)-PDK4 expression by phenylephrine and angiotensin II results in attenuated PDC activity, which is an attractive target for treating several metabolic dysfunctions including agonist-induced cardiac hypertrophy and diastolic dysfunction [224].
The increased glycolysis owing to sympathetic stimulation mediated by the mitochondrial Ca2+ influx affects PDP1 activity and is correlated with the activation of PDC following the glycolytic flux [25]. Interestingly, angiotensin type 1 receptor blockade using Irbesartan potentiates both palmitate oxidation and lactate oxidation compared to the control, along with the recovery of glucose oxidation, suggesting that more ATP production occurs via diverse metabolic pathways for better contractibility [24]. Mitochondrial 3-hydroxy-3-methyglutaryl-CoA synthase 2, the rate-limiting enzyme for ketone body synthesis, is increased under the decreased glucose uptake because of insulin deficiency, demonstrating metabolic adaptation by shifting the flux of excess intramitochondrial acetyl-CoA from β-oxidation into ketone body production, regulated by PDK4 overexpression [26]. DCA, an activator of PDC, exhibits the beneficial effect on hypoxia-induced contractile malfunction in male rainbow trout by increasing oxidative metabolism, lowering lactate production, and Ca2+ handling similar to cardio-protection in females, independent of female sex steroids [27]. The improved diastolic function in T2DM models by DCA treatment is confirmed concomitantly, by the increased 13C bicarbonate and decreased 13C alanine via the tracking of hyperpolarized [1-13C] pyruvate in DCA-treated diabetic cardiomyopathy in vivo [28]. The robust cardiac PDK4 expression is selectively degraded by ATP-dependent mitochondrial proteases (Lon) within approximately 1 hour, which is now being considered as a therapeutic target compared to the other PDKs [29]. According to tracer analysis of the radiolabeled metabolites, SIRT1 is required for rapid cardio-protective adaptation by stimulating glycolysis and inhibiting fatty acid-dependent respiration, resulting in increased glucose dependency [30]. In brief, therapeutic targets for heart failure induced by metabolic inflexibility should be effective for both glycemic control and to improve cardiac dysfunction, which is of great clinical significance.
Muscle
Heterogeneous skeletal muscle tissue consists of three distinct phenotypes; red slow oxidative (type I), red fast oxidative-glycolytic (type IIa), and white fast glycolytic (type IIb). They exhibit distinct phenotypes in terms of fuel selection, and can account for 80% of insulin-mediated glucose disposal in humans. PDC activity increases during the fed state, as PDK activity declines in skeletal muscles, which is lowest in the white fast glycolytic muscle type. On the other hand, during starvation, PDK4 expression levels increase, followed by a sharp decline in the PDH activity and pyruvate oxidation in both red and white skeletal muscle tissues in order to restrict further oxidation of glucose [31]. In response to starvation, adenosine monophosphate (AMP)-activated protein kinase (AMPK) initiates the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) signaling pathway, which is responsible for the rapid structural and metabolic adaptation from glucose to fatty acids [3233]. Furthermore, re-inactivation of PDH by PDK4 during post-exercise affects the shift from glucose oxidation to glycogen re-synthesis, in favor of higher gluconeogenic recycling of carbohydrate-derived substrates [34]. Meanwhile, the constitutive activation of PDC by genetically manipulated PDK2/4 knockout mice displays increased glucose oxidation, but also increased re-esterification of acyl-CoAs into diacylglycerol and triacylglycerol. This results in the inhibitory effect of protein kinase C θ (PKC-θ) on insulin signaling in muscles, which is a debate for the mechanistic importance of PDH activation [35].
In the Randle cycle, the attenuated PDC activity mediated by PDK plays a critical role in insulin resistance of skeletal muscles, which is also known as metabolic inflexibility determined by energy sources. The additional increase in PDK4 expression in response to saturated or monounsaturated fatty acids are exhibited differently in myotubes cultured from obese and T2DM patients, suggesting that the fundamental alterations to the expression patterns in the genome already occur due to prolonged exposure to fatty acids, causing metabolic imbalance [36]. The main target for the compound mangiferin and the natural NAD+ precursor nicotinamide riboside, also known as a SIRT activator, appears to be PDC activity, to elicit a fuel-switching effect from fatty acids to glucose demonstrated by increased (carboxy-14C) pyruvate oxidation into CO2 and a higher respiratory quotient ratio in skeletal muscle [37]. In addition, recovery of the decreased running distance because of muscle insufficiency requires the pharmacological activation of PDC by DCA, although muscle mass and power is recovered simply by a high protein diet, in the renal failure model of 5/6 nephrectomized mice [38]. Mice with amyotrophic lateral sclerosis carrying the Cu/Zn superoxide dismutase gene (SOD1G93A) showed an early metabolic shift from glucose to lipid metabolism due to PDK4 induction, causing metabolic imbalance and denervation in glycolytic muscle fibers, which were ameliorated by DCA administration [39].
Recently, miR-182, which is highly expressed in fast-twitch muscle, was shown to be downregulated by a high fat diet, which elicits translocated FoxO1-induced PDK4 overexpression leading to a metabolic switch from the utilization of glucose to lipids and metabolic inflexibility in obese mice [40]. In terms of age-related insulin insensitivity, the plasma lactate and insulin-stimulated dephosphorylated PDH levels are sensitive biomarkers in skeletal muscle biopsies of only young subjects, but not in sedentary aging subjects [41]. On the other hand, the ketone ester-based form of nutrition provides an alternative fuel for oxidative phosphorylation and improves physical performance in athletes by maximizing human metabolic power via the upregulation of muscle fat oxidation [42]. These studies highlight the importance of PDC activity in coordinating fuel selection to maintain metabolic flexibility in skeletal muscle. Taken together, the increased phosphorylation of PDH E1α positively correlated with its own inactivation may be derived from the increased lipid delivery, leading to excessive β-oxidation and causing impaired insulin action on glucose utilization as an energy source.
Liver
One of the physiological functions of the liver is to maintain the glucose level in the plasma to provide metabolic fuel in other tissues through gluconeogenesis or glycogenolysis. Under fed conditions, the liver stores excess glucose as glycogen and converts the fatty acids into triglycerol for export, with very low density lipoproteins. The reduced PDC-generated acetyl-CoA in the liver is a prerequisite for blockade of the de novo lipogenic pathway, which is simultaneously compensated by stimulating lipogenic genes in adipose tissue to maintain the hepatic acetyl-CoA content, independent of hyperinsulinemia with lower blood glucose in liver-specific PDC deficiency [43]. Generally, SIRTs 1-3 carry a robust lysine deacetylase domain, while SIRT 4-5 display little to no deacetylation activity, especially the mitochondrial SIRT4. SIRT4 has lipoamidase activity that is superior to its deacetylase activity, hydrolyzing the lipoamide cofactors from the E2 component dihydrolipoyllysine acetyltransferase to diminish PDC activity after metabolic flux switching by glutamine stimulation in hepatic cells [44].
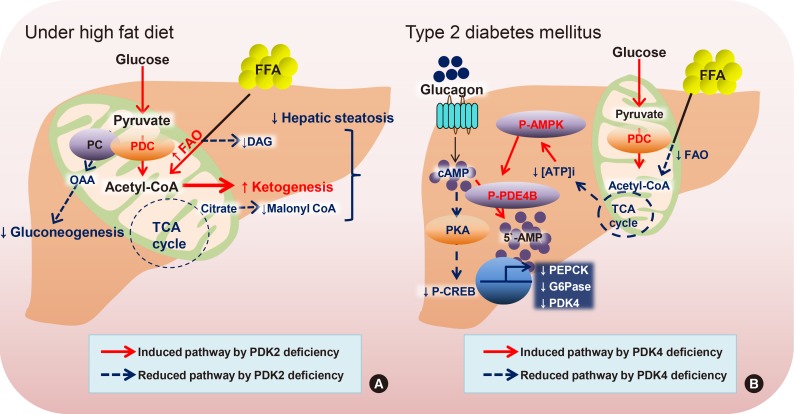
Based on a comparison between PDK2 and PDK4 gene knockouts under diabetic conditions (insulin receptor substrates 1/2 deficiency in vivo as a T2DM model), it was discovered that Pdk4-deficient mice exhibited better improvement in hyperglycemia and glucose tolerance, whereas Pdk2-deficient mice displayed higher insulin sensitivity, suggesting that hepatic Pdk4 would be a better clinical target for metabolic disturbances in T2DM [45]. Recent data from stable isotopomer flux (13C6-glucose) analysis showed that elevated PDK2 expression plays a major role in hepatic steatosis with insulin resistance, increased PC and TCA flux, decreased glucose oxidation, and decreased ketone body production. These symptoms are all ameliorated in Pdk2-deficient mouse models, confirmed by increased β-oxidation and ketogenesis because of reduced TCA anaplerosis by the re-activated PDC (Fig. 2A) [46]. A new drug candidate that acts as a PDK2-specific inhibitor by occupying the ATP-binding pocket was shown to ameliorate hepatic steatosis, consistent with drastically increased hepatic PDC activity, making it a great therapeutic target for obesity and T2DM [47]. Overall, hepatic steatosis is associated with mitochondrial oxidative alterations, which is related to an impairment of ATP synthesis under high-nutrient conditions and starvation, paradoxically. According to our unpublished data, hepatic PDK4 expression is increased in livers from fasted wild type mice or diabetic mice, both of which are represented by facilitated cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA)-cAMP response element binding protein (CREB) signaling. Inhibition of PDK4 in the liver reduces hepatic cAMP levels, and in turn, attenuates PKA-CREB signaling resulting in reduced gluconeogenesis. This was achieved by inhibition of rate of FAO by PDK4 deficiency. Given that FAO is responsible for ATP generation during gluconeogenesis, hepatic ATP level was drastically decreased in PDK4-deficient hepatocytes. This decrease was sufficient to increase phosphorylation of AMPK and subsequent phoshphodiesterase 4B (PDE4B); the latter responsible for the degradation of cAMP. Taken together, we suggest PDK4 as a potential target against diabetes where glucagon-cAMP-PKA-CREB signaling is aberrantly reinforced (Fig. 2B).
Brain
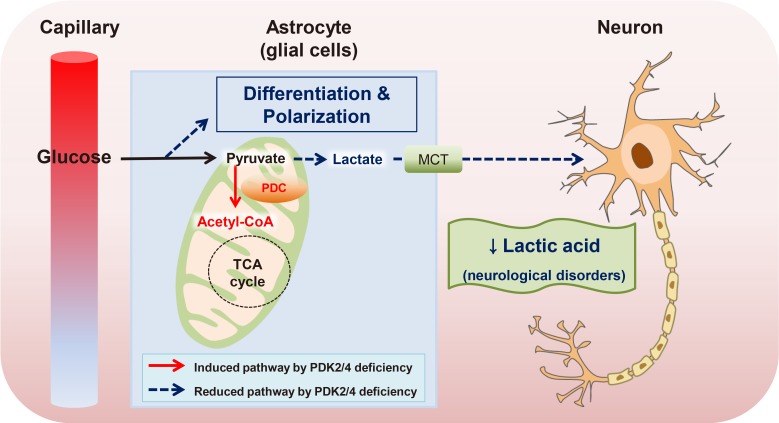
Lactate and alanine produced by astrocytes, a type of glial cell, are metabolized through the mitochondrial TCA cycle in neurons, which perform the energy-consuming transport of neurotransmitters. This metabolic interaction is proposed as the astrocyte-neuron lactate shuttle hypothesis [48]. The stimulated ketogenesis by 2-deoxy-D-glucose treatment lessens both amyloid precursor protein and amyloid-β oligomers by increasing the mitochondrial bioenergetics capacity, leading to the delay progression of Alzheimer's disease [49]. The metabolic modulation by ethanol and normobaric oxygen is mediated by increased PDC activity with the collaboration of increased PDP and decreased PDKs, which results in neuroprotection in severe stroke [50]. Likewise, PDC activity in astrocytes is strongly inhibited via phosphorylation by higher expression of PDK2 and PDK4 compared to neurons [51]. Deficiency of PDK2 and PDK4 in mice exhibits the attenuation of nociceptive behaviors in an inflammation-driven chronic pain model and in diabetic neuropathy, suggesting that lactic acidosis following anaerobic glycolysis is a critical step that is connected to metabolic reprogramming in the development of neurological disorders [52].
Similarly, PDC deficiency in a fetus causes congenital lactic acidosis and malfunctioning of the central nervous system, which can be rescued by a ketogenic dietary formula composed of high fat, adequate protein, and low carbohydrate [53]. Several identified mutants in human PDC have been inherited in peripheral neuropathy and in severe encephalopathy, characterized by increased lactate production, decreased oxidative phosphorylation, and defective mitochondrial homeostasis [5455]. A clinical study based on serum amino acid metabolite profiling demonstrates that metabolic dysfunction in myalgic encephalopathy/chronic fatigue syndrome is linked to defects in PDC activity, resulting in excessive lactate secretion, while the increased mitochondrial respiration utilizes the amino acids [56]. The complex electrical network in the brain is metabolically supported by the neuron-glia interaction. Therefore, the physiological mechanism of metabolic disturbances in the brain should be considered by looking at the distinct role of each cell type (Fig. 3).
Kidney
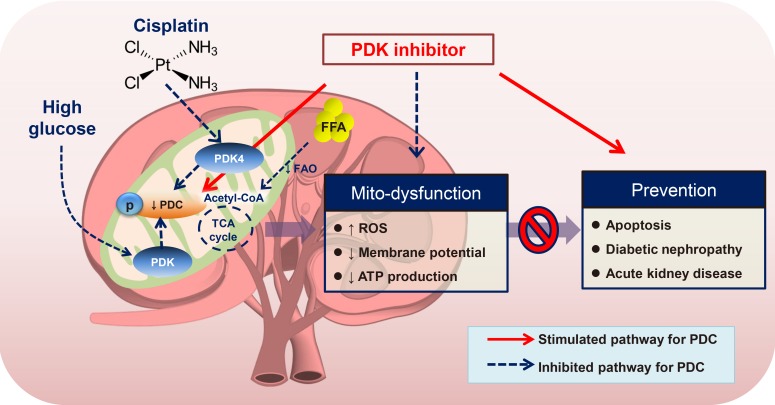
The kidney cortex comprises approximately 90% of proximal tubule and relies heavily on the oxidation of fatty acids compared to that of glucose for excretion of wastes, reabsorption of vital nutrients, acid-base homeostasis, osmolality regulation, and further blood pressure regulation. The reduction of mitochondrial biogenesis and increased PDH phosphorylation is observed in the streptozotocin-induced T1DM model, which is consistent with diminished AMPK activity, the major energy-sensing marker [57]. Cisplatin, one of the widely used chemotherapeutic agents elicits unwanted nephrotoxicity, which is mediated by PDK4 induction, leading to mitochondrial dysfunction and cellular apoptosis [58]. On the other hand, in spontaneously hypertensive rats, the increased mitochondrial oxidative phosphorylation in renal proximal tubule cells is attributed to higher PDC activity, which leads to hypertension [59]. Data from urinary metabolite analysis and in vivo metabolic flux analyses in diabetic complications show that the link between enhanced protein acetylation of metabolic enzymes and upregulation of urinary TCA cycle intermediates is seen in patients with nephropathy, not retinopathy [60]. Although flux studies using radiolabeled substrates for linking between metabolic and functional investigations have been introduced to nephrologists, further research is required to explore metabolic reprogramming in dysfunctional kidneys (Fig. 4). Briefly, we summarized the different mitochondrial PDC activities associated with metabolically important tissues (Table 1).
CONCLUSIONS
Inactivation of PDC helps the preservation of substrates such as pyruvate, lactate, and alanine for gluconeogenesis or cellular growth. Therefore, selective PDK inhibitors have been highlighted as novel anti-cancer agents to treat metabolic disturbances in cells, in addition to improving hyperglycemia. Metabolic reprogramming is a critical step for the individual cells regulated by epigenetic and post-modification mechanisms such as methylation, acetylation, and succinylation on metabolic genes. Taken together, the advanced systemic approach combining transcriptomics, metabolomics, and metabolic flux analysis enables us to explore metabolic pathways in a sophisticated way and reveal unexpected information beyond previous knowledge, supported by rapid progress in state-of-the-art technology. Consequently, a better and deeper understanding of the mechanisms underlying the metabolic alterations enables us to develop drugs that are more efficient with better selectivity and without unfavorable side effects, for use in patients suffering from metabolic diseases.
 XML Download
XML Download