

 PDF
PDF ePub
ePub Citation
Citation Print
Print



The Sulwon Award for Scientific Achievement is the Korean Diabetes Association's highest scientific award and honors an individual who has excellently contributed to the progress in the field of diabetes and metabolism. Sulwon Award is named after an emeritus professor Eung Jin Kim, who founded Korean Diabetes Association. Prof. Young Seol Kim received the ninth Sulwon Award at 2017 International Conference on Diabetes and Metabolism, September 28 to 30, 2017 at Seoul, Korea.
INTRODUCTION
Chronic hyperglycemia, a contributing factor in diabetic vascular complications, has been evaluated through experimental and large-scale clinical studies, such as the Diabetes Control and Complications Trial (DCCT) and the UK Prospective Diabetes Study (UKPDS) [123]. A follow-up DCCT study found that when the regulation of blood glucose was controlled strictly for 6.5 years at the initial stage of type 1 diabetes mellitus (T1DM), the effect lasted so long that the progression of microvascular complications was delayed for at least 8 years after the termination of the initial study, and the occurrence of cardiovascular events was delayed for 11 years [2]. According to the UKPDS follow-up study, patients with type 2 diabetes mellitus (T2DM) also had the legacy effect stemming from the strict regulation of blood sugar from the initial stage of the disease, which suggests that the phenomenon of metabolic memory exists in the vascular complications of diabetes [3].
Recent studies have suggested that advanced glycation end-products (AGEs) may be a key factor in the development of metabolic memory in diabetic complications, because AGEs are produced and accumulated irreversibly in the body, depending on the degree of blood sugar regulation and duration [4]. Once produced, the material is metabolized so slowly that it can well explain the phenomenon of metabolic memory or legacy effect [5]. Direct evidence of the relationship between the accumulation of AGEs and development of diabetic vascular complications was reported recently [6].
In this review, we summarized the recent AGE-related updates that have an important influence on the occurrence and deterioration of these diabetic vascular complications.
ADVANCED GLYCATION END-PRODUCTS
AGEs are produced by the Maillard reaction. A reducing sugar, such as glucose, reacts non-enzymatically with the amino group of proteins to produce a Schiff base that rearranges into Amadori compounds [5]. These Amadori adducts then very slowly undergo irreversible dehydration and condensation reactions, eventually producing AGEs, which are yellowish-brown materials with particular fluorescence [5].
AGEs are produced not only from glucose but also from the dicarbonyl compound produced from autoxidation and the degradation products of glucose, such as glyoxal, methylglyoxal, and 3-deoxyglucosone, or α-hydroxy aldehydes such as glyceraldehyde and glycolaldehyde [78]. In the case of chronic hyperglycemia, AGEs are actively produced and accumulate in the circulating blood and various tissues, resulting in vascular complications in diabetes [5].
AGE-RAGE SYSTEM IN DIABETIC VASCULAR COMPLICATIONS
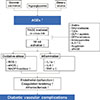
AGEs accelerate the expression of receptors for advanced glycation end-products (RAGEs). The constant activation of the AGE-RAGE system is presumed to create the long-term metabolic memory or legacy effect [11]. Upon the recognition of AGEs by RAGE in endothelial cells, the production of oxidative stress is accelerated in the cells, and various cytokines and growth factors are secreted through the activation of nuclear factor κB, thereby inducing the expression of adhesion factor and causing an inflammatory response (Fig. 1) [12]. In patients with diabetes, RAGE expression is accelerated in atherosclerotic lesions in proportion to aggravation of blood sugar regulation [13].
The oxidative stress accelerated by AGE-RAGE inactivates nitric oxide and then either causes an inflammatory response or aggravates a thrombotic tendency that leads to the progression of arteriosclerosis [1415]. In endothelial cells, the AGE-RAGE system promotes the gene expression of p22phox and gp91phox, which are components of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and accelerates the production of oxidative stress by activating the cell membrane transport of Rac family small GTPase 1 (Rac1) to cause endothelial cell dysfunction [16].
AGEs promote the production of vascular endothelial growth factor (VEGF) in the endothelial cells, which then induces pathological angiogenesis [17]. VEGF is known to be involved in the increasing atheroma in an atherosclerotic lesion [18]. This suggests that the AGE-RAGE system, through the production of VEGF, activates angiogenesis in atheroma and aggravates inflammation in the plaque, possibly by a hemorrhage or an increase in the atheroma [19].
In addition, the AGE-RAGE system inhibits the production of prostacyclin (PGI2) in endothelial cells and promotes the de novo synthesis of plasminogen activator inhibitor-1, thereby inhibiting fibrinolytic activity and then contributing to stabilization of the thrombus [19]. A decrease in cyclic adenosine monophosphate production through the AGE-RAGE system is expected to be involved in the thrombotic tendency [20].
AGEs are known to promote not only platelet aggregation but also the blood coagulation cascade through tissue factor production [21]. The thrombotic tendency induced by AGEs is considered the cause of acute coronary syndromes, such as unstable angina or acute myocardial infarction through atheroma rupture and subsequent thrombogenesis in the coronary artery [21].
The low density lipoproteins (LDLs) oxidized and denatured by AGEs are recognized by scavenger receptors, which then activate foam cell formation from macrophages and the secretion of chemokines or growth factors therein. The LDLs therefore accelerate the migration, proliferation, and transformation of smooth-muscle cells or the endothelial invasion by monocytes [22].
A clear progression of atherosclerosis and plaques accompanied by calcified lesions is observed in patients with diabetes [23]. In addition, the appearance of calcified lesions in atherosclerotic lesions is considered a predictive factor of future cardiovascular events and a marker of poor prognosis resistance to coronary intervention [23]. AGEs act on blood vessel wall cells, such as pericytes, promoting their differentiation into osteoblasts and thus causing calcification deposition [24]. Therefore, AGEs present in atherosclerotic lesions are presumed to be involved in the occurrence and progression of calcified lesions by acting on pericytes of the angiogenesis induced by an increase in atheroma [25].
AGE-RAGE SYSTEM WITH OTHER PATHWAYS
The oxidative stress accelerated by the AGE-RAGE system not only causes vascular disorder by activating the renin-angiotensin system (RAS) but also aggravates organ failure, because RAS activation causes NADPH oxidase-derived oxidative stress that again leads to an increase in RAGE expression as well as AGE production [2627]. This fact suggests that inhibition of the AGE-RAGE system is involved in some of the long-term protective effects of RAS inhibitors [27].
It is known that in hyperglycemia, the de novo synthesis of diacylglycerol is accelerated and protein kinase Cβ (PKCβ) is activated, and sorbitol or fructose is accumulated in the cells as a result of polyol pathway hyperactivity [28]. PKCβ activation is presumed to accelerate the activity of NADPH oxidase and then causes vascular complications in diabetes in connection with the AGE-RAGE system [28]. Fructose has a 10-time stronger glycation activity than glucose, and AGE formation from intracellular protein and fructose may be involved in vascular disorders [29]. The mitochondrial-derived oxidative stress produced by the hyperglycemic load causes AGE production, PKCβ activation, and polyol pathway hyperactivity [28]. These three hypotheses, which have existed for many years, and the AGE-RAGE system have a mutual role in the development of diabetic vascular complications [30].
AGE AND sRAGE AS INDICATORS OF VASCULAR COMPLICATIONS
Serum levels of AGEs are known to constitute an independent risk factor of endothelial dysfunction in patients with T2DM [4]. In addition, serum levels of AGEs were shown to be an independent factor for predicting death from cardiovascular events in women with T2DM or in nondiabetic patients in an 18-year follow-up study [30]. It is also known that serum AGE levels are related to left ventricular diastolic dysfunction and vascular stiffness in patients with T1DM [31].
RAGE has three splice variants of full-length RAGE, an N-terminal variant that does not contain an AGE-binding domain, a soluble receptor for advanced glycation end-product (sRAGE), and a C-terminal splice variant that does not include transmembrane and effector domains [32]. The physiological function of sRAGE has not yet been fully characterized, but it is known to be produced through cleavage of cell-surface RAGE and is known to reflect endothelial cell damage [33]. However, it is known that sRAGE can modulate the AGE-RAGE system as an antagonist of full-length RAGE [34].
Although serum levels of sRAGEs are only about 1/1,000 to 1/5,000 of the AGE concentrations in circulation, sRAGE could become an indicator for predicting the degree of vascular disorders because it reflects the RAGE expression in tissues [30]. One study also demonstrated that serum sRAGE levels were significantly higher in T2DM patients than in non-diabetic subjects and positively associated with the presence of coronary artery disease [33].
AGEs IN FOODS
Because the AGE reaction is also referred to as the browning reaction, the browning change of foods can be a measure of their AGE content. In general, it is known that a large amount of AGEs are produced when meats or fatty foods are heated or baked at a high temperature [35]. On the other hand, AGEs are not produced in a significant amount when foods are slowly steamed or heated with a lot of water for a long time [35]. For example, chicken shows a clear browning reaction, and grilled chicken skewers in particular have about 10 times higher levels of AGEs than chicken boiled in water [35].
Because assessments of their eating history and eating habits suggest a correlation between the amount of AGE intake and serum AGE level, a decrease in the amount of food-derived AGE intake is considered to inhibit vascular complications in diabetic patients [3536]. In this regard, some experiments also reported that the long-term consumption of AGE-limiting foods in mice reduced oxidative stress markers and inhibited insulin resistance and organ failure, resulting in an extension of life span [37]. Therefore, the limitation of food-derived AGEs can become a significant therapeutic target for inhibiting organ failure due to diabetes or aging.
Indeed, when patients with T2DM consumed AGE-restricted diet, the serum levels of AGEs decreased by around 30% to 40% in 2 to 6 weeks, resulting in a 20% decrease in markers of vascular complications, such as soluble vascular cell adhesion molecule-1 or C-reactive protein, or inflammatory markers [38]. In addition, it is known that in diabetic patients who limited food-derived AGEs, the AGE formation or oxidative degeneration of blood LDL-cholesterol is inhibited, and vascular complications due to LDL-cholesterol are also decreased [39]. However, it is not yet known whether the long-term, AGE-limiting diets can inhibit vascular complications in diabetic patients and improve their prognosis. In addition, differences in the absorption capacity of the gastrointestinal tract; the binding capacity with RAGE; and organ failure, depending on the type of AGEs taken up by the body, should be considered.
AGENTS KNOWN TO MODULATE AGEs
Several drugs are known to modulate AGEs (Table 1). Statin could slow the AGE-RAGE-mediated activation of NADPH oxidase by inhibiting the prenylation of Rac1, which subsequently inhibits post-RAGE-induced signal transduction [40]. Pravastatin is known to reduce tubular damage in diabetic nephropathy in tubular cells and attenuate AGEs-induced apoptosis [41]. Atorvastatin is also known to inhibit AGE formation through its anti-oxidative activity [42].
Telmisartan, an angiotensin receptor blocker with selective peroxisome proliferator-activated receptor γ activation, is known to inhibit RAGE expression in endothelial cells, renal mesangial cells, and hepatocytes [43]. It is known to inhibit the expression of oxidative stress and inflammatory markers by inhibiting signal transduction by AGEs and expression of arteriosclerosis-related genes [4344]. It is also known that ramipril, an angiotensin-converting enzyme inhibitor, can regulate AGEs with a similar mechanism of action [45].
The AGE modulatory potential is also known in some anti-diabetic medications. Rosiglitazone could reduce the expression of RAGE on the myocardium and attenuates cardiac fibrosis and ventricular diastolic function in experimental models [46]. Exendin-4 could inhibit the AGE-RAGE-mediated damage in tubular cells to attenuate the development and progression of diabetic nephropathy [47]. Linagliptin also has beneficial effects on diabetic nephropathy, partially inhibiting AGE-RAGE-evoked oxidative stress generation [4849].
Therefore, these agents are regarded as AGE-RAGE modulators in a broad sense. However, it is still unclear whether the modulation of AGEs by these drugs plays an important role in the improvement of diabetic macrovascular complications, compared to the main effects of these drugs.
Recently, the efficacy of aminoguanidine has been evaluated as a novel intervention for AGEs. Studies have shown that aminoguanidine inhibits the formation of AGEs and inhibits micro- and macrovascular complications in apolipoprotein knockout mice [505152]. It is also known that AGE cross-link breakers such as ALT-711 or alagebrium may help prevent diabetic vascular complications [4552]. In addition, the effect of direct antagonism on AGE of sRAGE has been demonstrated in some experimental studies [32]. However, the effect of the AGE modulator on advanced complications is unclear. In addition, there are few intervention studies of the actual clinical usefulness.
CONCLUSIONS
The formation of AGEs in diabetes patients occurs as a consequence of chronically elevated blood glucose levels. Some of the AGEs are known to be ingested through food. These AGEs could be one of the major contributing factors to the development of diabetic complications. It increases oxidative stress in the body according to various mechanisms and induces an inflammatory reaction, which has an important influence on the occurrence and exacerbation of diabetic vascular complications.
Recently, several clinical and preclinical studies have shown that interventions for these AGEs may play an important role in the reduction and prevention of diabetic vascular complications. This can be achieved not only by proper blood glucose control but also through diet and medical intervention. Recently, new compounds for the regulation of AGEs in the body have been studied.
In the future, further research results will clarify the role of AGEs in diabetic vascular complications, and we hope that successful intervention will help prevent complications and improve the quality of life for diabetic patients.

 XML Download
XML Download