

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Obesity has become a public health concern in many countries [1]; thus, many researchers are conducting studies to understand the pathogenesis of obesity and develop effective treatments. Obesity is a complex disorder accompanied by numerous changes in body composition and metabolic homeostasis. Although recent obesity epidemics can largely be explained by environmental changes, the development of obesity in individuals exposed to similar environments is affected by their genetics [2]. A study on monozygotic twins revealed that the degree of weight gain upon overeating was well-correlated between monozygotic twins [3]. On the other hand, obesity-related metabolic pathways have been discovered through genetic animal models and human genetic disorders that manifest obesity [45]. A representative example is leptin and leptin receptors. Severe obese phenotype observed in ob/ob mice and db/db mice led to the discovery of leptin and its functioning receptor leptin receptor long isoform (LepRb) [67].
Primary cilia were first discovered approximately 150 years previously and were thought to have no function or a very minor one for approximately 100 years. However, recent advances in science technology have provided new information on the structure and functions of cilia [8]. Therefore, these vestigial organelles are now regarded to function as signaling centers that have a pivotal role in transducing many signaling pathways [9]. Studies on genetic abnormalities in proteins located near primary cilia (so-called genetic ciliopathy) have helped us in understanding biochemical processes related to the primary cilia. These studies have also revealed crucial roles of primary cilia in sensing changes in extracellular environments and transducing this information into cells through diverse molecular signaling pathways [10]. Among these inherited ciliopathies, Bardet-Biedl syndrome (BBS) and Alström syndrome (ALMS) provide unique insights into cellular pathways involved in obesity and insulin resistance [4]. In this review, we provide a brief overview on the primary cilia and their roles in coordinating signal transduction in several metabolic organs and then review recent studies on the roles of primary cilia and cilia-related signaling in the development of obesity.
STRUCTURE OF PRIMARY CILIA AND THE INTRAFLAGELLAR TRANSPORT SYSTEM
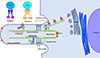
A primary cilium is a microtubule-based structure on the surface of a cell body that protrudes into the extracellular space [11]. Non-motile primary cilia are ubiquitously expressed in most vertebrate cells, with some exceptions such as dividing cells. Conversely, motile cilia and flagellae exist in specialized tissues. In addition, most cells have only one primary cilium, unlike multiple motile cilia [12]. A primary cilium comprises a central cytoskeleton core, called as an axoneme, which consists of nine pairs of microtubules, and a specialized cell membrane covering the axoneme (Fig. 1) [11]. The basal body, a structure that derives from the centrioles after cell division, anchors the primary cilium through transition fibers. Assembly of protein complexes transported to the primary cilium occurs around the basal body (Fig. 1) [12].
The plasma membrane of a primary cilium is enriched with many protein complexes, such as sensing and signaling receptors, ion channels, effector proteins, and transcription factors, that are necessary for signal reception, integration, and transduction [9]. Because primary cilia have no machinery to produce proteins, ciliary components are made externally and transported into the cilia through a specialized transport system and selective entry. Ciliary proteins containing a ciliary targeting sequence undergo post-translational modifications in the Golgi complex for proper targeting to the primary cilia and then migrate to the basal body along the microtubule network through polarized vesicle trafficking (Fig. 1). There are many substances and biochemical pathways involved in this system. Typical examples are small GTPases, such as Rab GTPases. Among these Rabs, Rab8 acts as a key mediator for vesicle trafficking to the cilia [13].
Upon arrival at the cilium base, axonemal precursors and ciliary membrane proteins are transported to the tip of the cilia by the intraflagellar transport (IFT) machinery (Fig. 1). IFT particles comprise 17 proteins that form two complexes: IFT complex A (IFT-A) and IFT complex B (IFT-B) [11]. During antegrade transport from the base to the tip of the primary cilia, cargo proteins bind to IFT-B with kinesin-2 that provides the power of motility and travel along the axoneme [14]. Retrograde transport from the tip of the cilia to the base is performed by IFT-A with a motor protein, dynein-2 [15]. Critical roles of IFT in the formation and function of the primary cilia have been elucidated by genetic studies. For example, genetic abnormalities in IFT88 (a component of IFT-B), IFT122 (a component of IFT-A), and KIF3A (a subunit of kinesin-2) cause absence or shortening of the cilia and abnormal signal transduction [161718].
SIGNALING PATHWAYS RELATED TO PRIMARY CILIA
Thus far, >15 signaling transduction pathways are known to be related to the primary cilia. Therefore, a primary cilium is regarded as a hub for signal transduction [4]. The Hedgehog (HH) signaling pathway is the most well-known signaling system that is coordinated by the primary cilia. The HH signaling pathway has a crucial role in the maintenance of tissue homeostasis and regulation of cell differentiation during embryonic development. Recent studies have also highlighted the role of HH signaling in the regulation of adult stem cells and development of some cancers [19]. Therefore, drugs targeting HH signaling to fight cancers are being actively developed by many pharmaceutical companies.
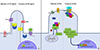
HH molecules are polypeptide ligands that initiate the HH signaling pathway. HH was named after the HH-like morphology of HH-mutant drosophila [20]. Mammals have three HH homologs: desert (DHH), Indian (IHH), and sonic (SHH). Of these, SHH is the widely studied [212223]. Upon binding of HH to the Frizzled-type receptor patched, G protein coupled receptor (GPCR)-like protein smoothened (SMO) accumulates in cilia (Fig. 2A). Inside the cilium, SMO induces downstream signaling that includes dissociation of GLI proteins (transcriptional effectors of the HH pathway) from the key HH pathway regulator suppressor of fused (SUFU). Dissociation of GLI and SUFU blocks GLI processing to give repressor forms and results in accumulation of GLI in cilia. Therefore, active-form GLI proteins traffic to the nucleus and activate GLI-dependent transcription (Fig. 2A) [24]. HH isoforms can also induce cellular responses that are independent of GLI-mediated transcriptional changes [25]. This non-canonical HH signaling includes Ca2+ and adenosine monophosphate (AMP)-activated protein kinase (AMPK) signaling [26].
Wnt signaling pathways are a network of signal transduction pathways which are highly conserved across species. Primary cilia have machinery associated with the Wnt signaling complex in the axoneme and regulate the balance of this signaling system. Wnt signaling regulates cell fate determination, proliferation, and migration during embryonic development; it is also partly involved in various insulin signaling pathways, such as sterol regulatory element-binding protein 1c (SREBP-1c), Akt, AMPK, and mitogen-activated protein kinase (MAPK); and thereby it enhances insulin sensitivity. In the central nervous system, leptin can activate Wnt signaling in orexigenic neuropeptide Y (NPY) neurons by inhibiting glycogen synthase kinase-3β (GSK-3β), thereby transmitting a catabolic action [27].
Wnt signaling is divided into canonical and non-canonical pathways depending on the involvement of β-catenin [28]. In the absence of Wnt ligands, β-catenin is ubiquitinated by a destruction complex (composed of axin, casein kinase-1α, adenomatosis polyposis coli, protein phosphatase 2A, and GSK-3β) and degraded in the proteasome (Fig. 2B). Upon binding of the Wnt ligand to the Frizzled family receptor and its coreceptors low density lipoprotein receptor-related protein-5/6 (LRP-5/6), the Frizzled receptor transmits the signal to the cytoplasmic phosphoprotein Dishevelled (Dsh). Frizzled receptor inactivates axin, and Dsh inhibits GSK-3β. These events lead to inhibited β-catenin degradation and increased β-catenin levels in the cytoplasm [29]. β-Catenin translocates to the nucleus and regulates target gene expression via the T-cell-specific transcription factor/lymphoid enhancer-binding factor 1 [30]. Non-canonical Wnt pathways are mediated by some domains of Dsh and use neither β-catenin nor LRP-5/6 coreceptors. Non-canonical Wnt pathways are involved in the regulation of the cytoskeleton, planar cell polarity, and intracellular Ca2+ levels by regulating calcium release from the endoplasmic reticulum [31].
Ciliary defect in kidney tubular epithelial cells causes a common genetic disorder called polycystic kidney disease (PKD). The cilium normally senses urinary flow to inhibit the proliferation of tubular epithelial cells through liver kinase B1 (LKB1)- and AMPK-mediated suppression of mammalian target of rapamycin (mTOR) signaling. LKB1 and AMPK are localized to the cilia-basal body in renal epithelial cells. Defective ciliary signaling in PKD leads to activation of the mTOR pathway, which triggers proliferation of epithelial cells and cyst formation [32]. The anti-diabetic drug metformin and mTOR inhibitor rapamycin have been shown to arrest cyst epithelial cell proliferation and suppress cyst growth [3233].
In addition to an important role of primary cilia in sensing and transducing external signals, cilia are also involved in transduction of internal signals. Recent studies have shown an intimate interplay between primary cilia and autophagy, a highly conserved intracellular process for degradation of proteins and organelles in lysosomes [34]. Autophagy is critical for cellular homeostasis as it removes damaged components and provides the cell with a new energy source for recovery of homeostasis in energy-deficient condition. Inducible autophagy in serum-deprived and energy-deficient conditions can promote ciliogenesis by degrading the oral-facial-digital syndrome 1 (OFD1) protein at the centriolar satellites, which act to inhibit ciliogenesis [35]. Conversely, basal autophagic activity during normal nutritional conditions can inhibit primary cilia growth by removing IFT components. Therefore, autophagic activity can differently regulate ciliogenesis depending on the nutritional context. On the other hand, primary cilia and HH signaling control autophagic activity. The molecular machinery involved in ciliogenesis also participates in the early steps of the autophagic process. Moreover, blockage of ciliogenesis by deletion of IFT20 and IFT88 reduces the autophagic activity [36]. These findings suggest that normal ciliary function is necessary for induction of autophagy.
PRIMARY CILIA AND HUMAN OBESITY
Ciliary dysfunction causes clinical syndromes with multi-organ developmental abnormalities termed as ciliopathies. Rare genetic disorders, such as BBS, ALMS, Joubert syndrome, and Meckel syndrome, result from mutations of genes associated with the cilium-basal body complex [10]. BBS is characterized by obesity, polydactyly, mental retardation, retinal degeneration, and renal and gonadal malformations. Thus far, 22 BBS proteins have been identified. Among them, eight BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, and BBS10) form a protein complex called the BBSome, which is a component of the basal body and is involved in trafficking vesicles to the primary cilium [37]. Another ciliopathy ALMS that is caused by mutations in the ALMS1 gene, is also associated with obesity [38]. Nearly all patients with ALMS and more than half of the patients with BBS have been reported to be obese or overweight.
On the other hand, common polymorphisms in the first intron of the fat mass and obesity-associated (FTO) gene are associated with human obesity [39]. A regulatory element within the first intron of the FTO gene controls expression of ciliary protein retinitis pigmentosa GTPase regulator-interacting protein-1 like (RPGRIP1L), which is located in the transition zone of the primary cilium. Mice with hypomorphic RPGRIP1L have been shown to be hyperphagic and obese and show reduced leptin signaling in the hypothalamus. Moreover, RPGRIP1L appeared to mediate trafficking of the leptin receptor in the vicinity of primary cilia [40]. Therefore, FTO gene-associated obesity might be related to the primary cilia-basal body complex.
ROLE OF PRIMARY CILIA AND CILIA-RELATED SIGNALING IN CENTRAL REGULATION OF ENERGY METABOLISM
The hypothalamus is a brain region that has an important role in the regulation of food intake and energy balance. Thus, destruction of the mediobasal hypothalamus leads to obesity and hyperphagia [41]. The hypothalamic arcuate nucleus (ARC) is adjacent to the median eminence and surrounds the third ventricle. Thus, peripheral metabolic hormones and nutrients in the systemic circulation and cerebrospinal fluid can easily access ARC. ARC contains a group of neurons producing orexigenic NPY and agouti-related protein and another group of neurons producing anorexigenic proopiomelanocortin (POMC) [42]. These neurons primarily sense systemic metabolic alterations for controlling food intake and energy expenditure.
These ARC neurons have relatively long (average length, approximately 5 to 6 µm) primary cilia [443]. We and others have demonstrated that primary cilia in hypothalamic neurons are important for sensing metabolic signals and maintaining energy balance. Mice with ciliary dysgenesis in POMC neurons are obese [44]. Consistently, artificial induction of short cilia in the hypothalamic ARC of adult mice, by injecting small inhibitory RNA (siRNA) specific to Kif3a or Ift88 into ARC, has increased food intake but decreased energy expenditure, which leads to weight gain. Anorexic responses to leptin, insulin, and glucose are also reduced in mice with siRNA-mediated Kif3a or Ift88 knockdown in ARC [45]. These findings support the statement that hypothalamic neuronal cilia are important for regulation of energy homeostasis.
Conversely, genetic- or diet-induced obesity causes changes in ciliary lengths of hypothalamic neurons. The average cilia length was reduced in the hypothalami of diet-induced obese (DIO) mice. Similar to the effect in DIO mice, the length of hypothalamic neuron cilia was reduced in leptin-deficient ob/ob mice and leptin receptor-deficient db/db mice. These ciliary changes were not observed in the neurons of the hippocampus. The short cilia phenotype in ob/ob mice was rescued by 7-day leptin administrations. Therefore, obesity-associated changes appeared to be specific to hypothalamic neurons and hypothalamic cilia lengths are dynamically regulated depending on metabolic conditions during adulthood.
Leptin, a representative adiposity signal, is known to play an important role in the regulation of energy balance through activation of catabolic POMC pathways [46]. The functional leptin receptor LepRb has high expression in several regions of the brain, including the hypothalamus [7]. Of the several brain regions, hypothalamic ARC is an important area that mediates leptin's modulating actions on food intake and locomotion. Bbs2−/−, Bbs4−/−, and Bbs6−/− mice were hyperphagic, had low locomotor activity, and exhibited hyperleptinemia and increased leptin resistance. In these BBS mutant mice, the anorexigenic effect of exogenous leptin was attenuated and hypothalamic POMC expression was decreased [47]. Leptin resistance lasted in Bbs mutant mice even after their increased body weight and leptin levels were normalized by caloric restriction. These findings suggested that leptin resistance observed in BBS-deficient mice may not be the secondary effect of obesity and obesity-related hyperleptinemia. However, another study has shown that preobese Bbs4−/− mice have normal response to leptin in terms of feeding and hypothalamic Stat3 activation [48]. Based on this data, they insisted that cilia are not directly involved in leptin responses and a defect in the leptin signaling axis is not the initiating event leading to hyperphagia and obesity associated with cilia dysfunction. Therefore, further studies are needed to clarify the roles of primary cilia and BBS proteins in leptin signaling.
Molecular mechanisms underlying leptin resistance observed in Bbs mutant mice have been investigated. It was shown that BBS1 protein interacted with the leptin receptor and was involved in leptin receptor trafficking to the cell membrane. Abnormal perinuclear accumulation of leptin receptor was observed in Bbs1-deficient cells [49]. Melanin-concentrating hormone (MCH) is an appetite-stimulating neuropeptide produced in neurons in the lateral hypothalamic area. Ciliary localization of the MCH receptor 1 (MCHR1) is also disrupted in neurons from mice lacking the Bbs2 or Bbs4 gene. Moreover, BBS2 and BBS4 re-expression in cultured Bbs2−/− and Bbs4−/− hippocampal neurons restored Mchr1 ciliary localization. Mislocation of MCHR1 to cilia may provide a potential molecular mechanism to link ciliary defects to obesity [50].
CILIA-RELATED SIGNALING FOR CONTROL OF ADIPOGENESIS
The adipose tissue contains adipocytes that store surplus energy as a lipid and secrete biologically active substances called adipokine [51]. Adipocytes are differentiated from preadipocytes that have originated from mesenchymal stem cells [52]. Proliferating preadipocytes or mature adipocytes are non-ciliated However, transmission electron microscopy has revealed the transient presence of primary cilia during differentiation of cultured preadipocytes. SiRNA-mediated inhibition of Bbs10 and Bbs12 expression in differentiating preadipocytes impaired ciliogenesis and simultaneously activated adipogenesis signals through the GSK-3β pathway and nuclear accumulation of peroxisome proliferator-activated receptor γ (PPAR-γ) [53]. Moreover, increased triglyceride content has been observed in dermal fibroblast-derived adipocytes from patients with BBS [53]. These findings strongly suggest that increased adipogenesis may contribute to the development of obesity in subjects with BBS.
Wnt and HH pathways are known as potent inhibitors of adipogenesis. Wnt signaling functions as a molecular switch in regulating adipogenesis and maintains preadipocytes in an undifferentiated state. Inhibition of Wnt signaling causes spontaneous adipogenesis [5455]. HH signaling also seems to have a role in regulating nutrient storage. In fries, activation of the HH pathway in the fat body, an organ that functions like adipocytes and the liver in mammals, decreases fat formation, whereas fat body-specific inhibition of the HH pathway stimulates fat formation. Similarly, HH signals inhibit mammalian adipogenesis. HH treatment has been shown to reduce triglyceride accumulation in 3T3-L1 adipocytes. Conversely, 3T3-L1 adipocytes were increased when HH signaling was inhibited by expression of a dominant-negative form of the HH transcription factor GLI2 [56]. In line with this, mutant mice with fat-specific HH-activation displayed loss of the white fat tissue without changes in brown fat tissue mass. Treatment with SHH and HH activator smoothened agonist (SAG) blocked adipocyte differentiation in 3T3-L1 adipocytes, but had no effect on the differentiation of HIB-1B brown adipocytes [57]. Taken together, these in vivo and in vitro data suggest that activation of HH signaling blocks adipogenesis through inhibition of white adipocyte differentiation.
CILIA-RELATED SIGNALING IN THE CONTROL OF GLUCOSE METABOLISM
HH signaling is closely associated with glucose metabolism. HH partial agonism reprograms glucose metabolism toward aerobic glycolysis in adipocytes. Treatment of 3T3-L1 adipocytes with the HH signal stimulator SAG and SHH increased cellular glucose uptake, lactate production, and cellular NAD+/NADH ratio; in contrast, it reduced cellular oxygen consumption, which suggests increased aerobic glycolysis (the so-called Warburg-like phenomenon in cancers cells) [58]. SAG and SHH treatment rapidly increased phosphorylation of pyruvate dehydrogenase alpha-1, AMPK, and pyruvate kinases M1/M2. Moreover, reduced expression of these proteins blunted SAG-induced changes in glucose metabolism, which confirmed a critical involvement of these molecules in HH-promoted aerobic glycolysis. Indeed, these three proteins have been known to have key roles in determining the fate of glycolytic substrates and have been implicated in the Warburg effect and cancer cell growth.
Effects of HH agonists on cellular glucose metabolism were abolished in Smo−/− cells, indicating them to be SMO-dependent effects. SAG-induced glucose uptake was mediated by glucose transporter-4. However, it did not require glucose transporter 1 (GLUT1) and insulin receptor, which is similar to the characteristics of AMPK-driven glucose uptake. SMO activates GLI-dependent transcriptional regulation in hours or days [58]. In addition, SMO is a class F G-protein-coupled receptor [5960] and can activate G-protein-coupled second messenger signals in a few seconds or minutes. SMO-induced AMPK activation and metabolic reprograming was dependent on AMPK-upstream kinases LKB1 and calcium/calmodulin-dependent protein kinase kinase (CAMKK2). HH activators did not induce alterations in the cellular AMP/adenosine triphosphate (ATP) ratio. Instead, SAG treatment rapidly induced calcium influx via Gαi-dependent opening of the plasma membrane L-type calcium channels, which led to activated CAMKK2-AMPK signaling.
Canonical HH signaling requires SMO translocation to the primary cilium. Ciliary proteins IFT88 and KIF3A are required for proper ciliogenesis, and their loss results in lack of canonical HH signaling competency and functional primary cilium. Notably, in Ift88−/− and Kif3a−/− mouse embryonic fibroblast cells, SAG-induced glucose uptake and lactate production were severely blunted. Moreover, SAG-induced AMPK activation was also abolished, although SAG normally elicited a Ca2+ response. These results strongly indicated that the cilium is not needed for Ca2+ response to HH agonists, but it is critical for coupling of SMO-induction to AMPK activation. Contrary to expectation, treatment of 3T3-L1 cells with the canonical HH antagonist and partial SMO agonist cyclopamine and GDC-0449 induced the same Warburg-like effects triggered by the SMO agonist SAG. Mice receiving cyclopamine displayed a significant improvement in glucose tolerance. Cyclopamine infusion acutely increased glucose uptake in the brown adipose tissue and muscles, but not in the white adipose tissue, in an insulin-independent manner and led to improved glucose tolerance and enhanced thermogenesis [58]. Stimulation of myotubes or primary brown adipocytes with cyclopamine activated AMPK signaling and induced Warburg-like effects, as observed in 3T3-L1 cells. Therefore, in contrast to canonical HH signaling, which inhibits differentiation of the white adipose tissue to limit glucose metabolism, non-canonical HH signaling stimulates glucose and energy metabolism in skeletal muscle and brown adipocytes.
CONCLUSIONS
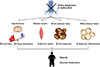
In this review, we discussed recent evidence suggesting emerging roles of primary cilia and cilia-mediated signaling pathways in the regulation of metabolism. Primary cilia constitute a restricted and narrow compartment and a wide surface that is in contact with the extracellular environment. Therefore, they can efficiently sense changes in the external environment and initiate signaling cascades. Defective or absent cilia in developing neurons and adipose stem cells led to obesity in humans and rodents (Fig. 3). The mechanisms underlying cilia-related obesity are now being unveiled. In hypothalamic neurons, primary cilia appear to act as a signaling platform conveying metabolic information. In the adipose tissue, cilia control the process of adipocyte differentiation by transducing canonical HH signaling and Wnt signaling. Moreover, non-canonical HH signaling in the skeletal muscle and brown adipocytes controls glucose metabolism, and these effects involve the activation of CAMKK2-AMPK signaling and cilia-related GPCR SMO. Therefore, cilia and cilia-related signaling molecules will be a potential target for new treatments combating obesity and type 2 diabetes mellitus.
 XML Download
XML Download