

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Shigellosis is an acute inflammatory bowel disease caused by a group of Gram-negative intracellular enterobacteria known as Shigella. Food and water sources that contaminated with human or animal waste contribute primarily towards the transmission of Shigella [1]. Based on the structure of their lipopolysaccharide O-antigen, the bacteria are divided into four species namely Shigella flexneri, S. sonnei, S. dysenteriae, and S. boydii [2]. At present, shigellosis has led to 169 million morbidities in developing countries and is responsible for 1.1 million deaths per year in young age children who are less than 5 years of age, attributed to Shigella-associated diarrhoea cases [3]. Apart from young children, Shigella also targets elderly and immunocompromised patients [4]. Its mode of transmission is by fecal-oral route and requires a minimum infectious dose of <10 to 100 bacilli to cause the infection, due to its ability to survive gastric acidity better than other enterobacteria [5]. In the context of pathogenesis, Shigella sp. invades and colonizes the colonic mucosa leading to its disruption. The bacilli invade the villi of the large intestine, multiply and spread laterally to adjacent epithelial cells and also penetrate into the lamina propria [5]. Since the pathogen is intracellular it becomes even more difficult to treat the disease. The symptoms associated with shigellosis range from mild self-limited diarrhea to severe dysentery with frequent passage of blood and mucus, high fever, abdominal cramps, malaise, chills, nausea, vomiting and in rare cases known to cause bacteremia.
The uncontrolled use of antimicrobial agents Shigella sp. has inadvertently led to the development of resistant strains, which hinder appropriate selection of effective antibiotics, causing it poses a threat to public health [6]. Shigella strains have been reported to develop resistance to sulphonamide followed by with an increasing rate of resistance to tetracycline, chloramphenicol, ampicillin, nalidixic acid, and fluoroquinolones, making therapeutic treatment against Shigella infection a challenge [6]. In Malaysia, literature study strongly suggests that Shigella spp. exhibits frequencies of resistance against tetracycline and trimethoprim-sulfamethoxazole. The existing antibiotics can no longer be used to treat cases of severe diarrhoea and dysentery [5]. Therefore, there is an urgent need for vaccine development for the long-term control of Shigella transmission in affected tropical regions.
Until now, no practical vaccine available for shigellosis has yet been licensed. Current existing vaccine candidates are either not sufficiently attenuated or less immunogenic to the host [7]. Further identification of immunogenic protective antigens is essentially required to develop effective subunit vaccines for shigellosis. Recent literatures reveal that the outer membrane proteins (OMPs) of Shigella are ideal targets for vaccine development [8]. It has been well established that the OMPs of Shigella are capable of induction of immunological response in animal models given these proteins play crucial role as dynamic interface during host-pathogen interaction [9]. In most of the bacteria, OMPs involve in cell structure maintenance, providing cell's integrity, maintaining selective permeable of bacterial membrane, an adaptation of bacteria in the host niches, and bacterial pathogenesis through enhancing the bacterial adaptability in various type of environment. Most of the OMPs are multifaceted surface exposed antigens which play an important role in outer membrane structure integrity and represent a virulence factor among various Shigella strains [10].
Development of vaccine through conventional techniques takes decades to develop. With the availability of whole genome sequences, the genomic data could be used in silico to help identify and screen for potential vaccine candidates. This vaccine development technique using genome-based in silico method is known as “reverse vaccinology” [11]. To select a potential vaccine candidate, it is essential to identify the virulent protein that is capable to evoke an immune response within the host organism. Some features required for an effective vaccine candidate protein include: (1) sub-cellular localization; (2) presence of a signal peptide; (3) transmembrane domain; and (4) antigenic epitopes. The main strategy behind identification of potential vaccine candidates is recognizing the antigenic and virulence factor as well as predicting those sequences which are likely to bind to major histocompatibility complex (human leukocyte antigen [HLA] in human) molecules on the antigen presenting cells within the host. Reverse vaccinology approach was firstly adopted by Rappuoli and his colleagues to develop a safe and protective vaccine against Group B meningococcus [12].
This study is based on the use of reverse vaccinology approach for the identification and search of putative vaccine targets against S. flexneri 2a. The most conserved and immunogenic OMPs of S. flexneri 2a which can be potent vaccine candidates were initially identified. The shortlisted proteins were then predicted for their antigenicity as potential epitopic sequences using validated online immunoinformatics software. A schematic diagram showing computational pipeline corresponds to reverse vaccinology used in this study is represented in Fig. 1.
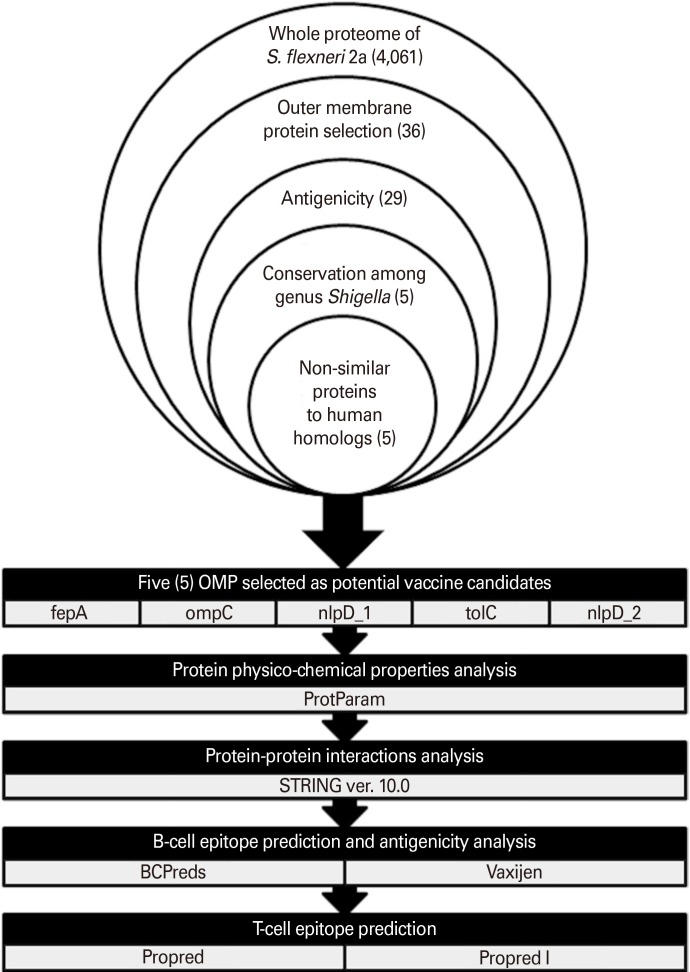
Go to : 
Materials and Methods
Data retrieval and proteome analysis
Primary proteomic data of Shigella spp. was retrieved from GenBank listed in the Vaxign program (http://www.violinet.org/vaxign/) documented in September 2017. The S. flexneri 2a strain 2457T genome was used as the seed genome in this study. The other Shigella genomes which were available in the Vaxign program include two sequenced virulent strains from main pathogenic Shigella species: S. boydii CDC and S. dysentriae SD197 were used for orthologues comparison.
Selection of outer membrane proteins
Vaxign was used for vaccine target prediction and analysis. This is a web-based program which contains a computational pipeline designed specifically for vaccine target prediction and analysis system based on the principle of reverse vaccinology [13]. The potential vaccine candidates are preselected according to their subcellular localization, probability of having adhesion-like characteristics, and number of transmembrane helixes. Briefly, subcellular localization is predicted using PSORTb ver. 2.0 (https://www.psort.org/) [14]. The default setting includes (1) cell wall, (2) cytoplasmic, (3) cytoplasmic membrane, (4) extracellular, (5) outer membrane, (6) periplasmic, and (7) unknown but only outer membrane-localized proteins were chosen in this study. Characteristic of transmembrane helices in proteins is predicted using TMHMM based on hidden Markov model [15], with the default value set to 1. The probability of adhesins and adhesin-like proteins is predicted by SPAAN software with the default cutoff is 0.51 [16].
Antigenicity analysis
Antigenicity of each pre-selected OMP in this study was computed using the Vaxijen ver. 2.0 server (The Edward Jenner Institute for Vaccine Research, Compton, UK). Vaxijen ver. 2.0 is an alignment-free approach for antigen prediction, which is based on auto cross covariance transformation of protein sequences into uniform vectors of principal amino acid properties. To increase the stringency of the prediction, the threshold value was set to 0.5%. Any protein that had an antigenic score above 0.5% was selected for further analysis [17].
Identification of conserved identity with other Shigella strains
In order to assess conservation of the selected proteins in the different bacteria strains, BLASTp analysis was performed for each amino acid sequence against S. boydii, S. dysentery, and S. sonnei whole genomes available on the NCBI server (National Center for Biotechnology Information, Bethesda, MD, USA). The identity percentage was set to 80% and the minimum query coverage was set to 50%. The amount and percentage of sharing among the available genomes were determined. The proteins with a sharing percentage <80% were considered not conserved proteins and were eliminated from the dataset.
Identification of human homologs
The predicted conserved OMPs of genus Shigella were subjected to BLASTp analysis to perform the similarity search against human proteome. The proteins having no significant identity (<35%) were considered to be sufficiently distant to the human proteome and will not interfere with normal host immune mechanism when used as a vaccine candidate [18].
Protein physico-chemical analysis
The location of signal peptide within the selected OMPs was analyzed using SignalP ver. 4.1 server (http://www.cbs.dtu.dk/services/SignalP/) [19]. The molecular weight and theoretical isoelectric point of the selected OMPs were computed using ProtParam server (https://web.expasy.org/protparam/) [20].
Protein-protein interactions analysis
In order to explore the functional pathway and interaction network of the OMPs having potential role in the vaccine development against S. flexneri 2a, protein-protein interactions of those selected proteins were analyzed using STRING ver. 10.5 (https://string-db.org/). Interactors having medium confidence score <0.700 were removed from the network to avoid false positives prediction [21].
B-cell epitope prediction
Immunogenic OMPs that conserved among the three Shigella strains were analyzed for their potential B-cell epitopes using BCPreds software (https://omictools.com/bcpreds-tool). BCPreds contains two methods based on different algorithms which are known as amino acid pair antigenicity method and BCPreds method using subsequence kernel [22]. All predicted B-cell epitopes (20 mers) having a BCPreds cutoff score >0.80 were selected. The B-cell epitopes resulted from the two algorithms were assembled and the overlapping regions were selected as predicted B-cell epitopes [23]. Selected B-cell epitopes were subsequently calculated for their antigenicity using VaxiJen ver. 2.0 server.
T-cell epitope prediction
Provided epitopes triggering both B-cell and T-cell immune responses are crucial targets for the development of diagnostics and therapeutics, nominated B-cell epitopes were then evaluated for their effective binding to HLA class I and HLA class II alleles using Propred (http://crdd.osdd.net/raghava/propred/) [24] and Propred I servers [25]. All the alleles from the list of 51 HLA class I and 36 HLA class II were exploited for the prediction of T-cell epitopes. B-cell epitopes having promiscuous T-cell epitopes (epitope presented by more than one HLA allele) binding to at least 15 HLA class I and class II alleles were listed and predicted as “binder.”
Go to :
Results
Data retrieval and proteome analysis
Using the Vaxign program, a total of 4,956 proteins representing the total proteome of S. flexneri 2a deposited in Genbank was retrieved. Subcellular localization of the proteins was predicted using PSORTb ver. 2.0. As per the predictions, 2,637 (54%), 896 (18%), and 135 (1%) of the total proteins are localized in cytoplasm, cytoplasmic membrane, and periplasm, respectively. Of the total proteins, 71 (1%) accounted for the outer membrane, whereas 37 proteins (1%) were classified as secretomes. Cell wall proteins were not found in S. flexneri 2a and other Shigella strains. Of these, 23% of the proteins were not identified and reported as unknown. The results of subcellular localization analysis are given in Fig. 2.
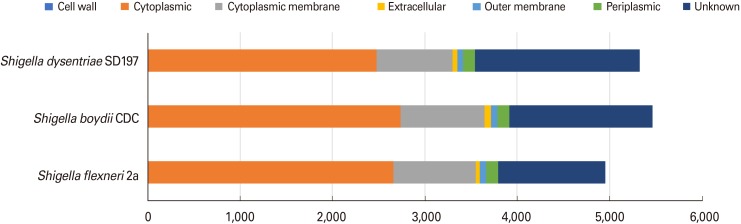
Selection of outer membrane proteins
Seventy-one proteins which have been classified as OMPs in the previous step were subjected to transmembrane helices and adhesion features analysis. Based on the selection criteria, 36 OMPs were selected as potential vaccine antigens. In order to understand the protein functional roles, all these OMPs of S. flexneri 2a were further analyzed using Clusters of Orthologous Groups of proteins database [26] available on Vaxign program. The OMPs were classified into 11 functional categories. Majority of the identified OMPs were predicted to be responsible for (1) carbohydrate transport and metabolism; (2) coenzyme transport and metabolism; (3) lipid transport and metabolism; (4) cell wall/membrane/envelope biogenesis; (5) cell wall/membrane/envelope biogenesis, inorganic ion transport and metabolism; (6) cell wall/membrane/envelope biogenesis, intracellular trafficking, secretion, and vesicular transport; (7) cell motility, intracellular trafficking, secretion, and vesicular transport; and (8) inorganic ion transport and metabolism. The classification of S. flexneri 2a selected OMPs according to functional categories is shown in Fig. 3.
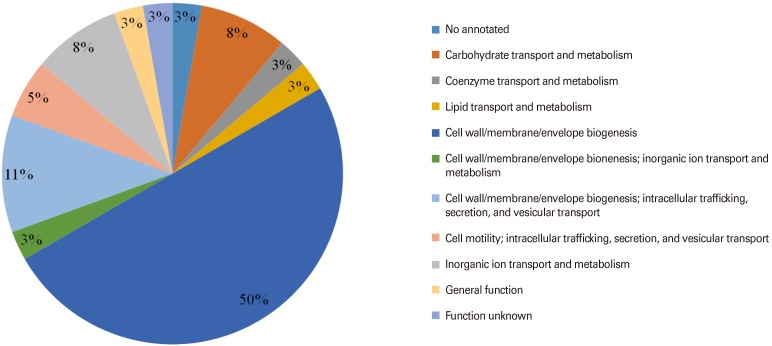
Antigenicity analysis
In order to refine the selection, the antigenicity of the 36 OMPs selected was calculated using Vaxijen ver. 2.0. Of these, 29 OMPs were found to have antigenicity score above the threshold value of 0.5 (antigenic), ranging from 0.51 (hypothetical protein S1556) to 0.85 (outer membrane fluffing protein). This step allowed eliminating seven OMPs with the antigenicity score lower than 0.5 which were considered as non-antigen.
Identification of conserved identity with other Shigella strains
In order to identify antigens which can provide cross-protection among Shigella species, BLASTp analysis was performed to assess the individual sharing of the selected OMPs among S. flexneri 2a with another two Shigella strains which are S. boydii and S. dysentery. The alignment showed that five OMPs from S. flexneri 2a shared great sequence identity (93%–100%) with S. boydii and S. dysentery (Table 1). Five of these highly conserved antigenic OMPs were outer membrane receptor (fepA), outer membrane porin protein C (ompC), lipoprotein NlpD (nlpD_1), outer membrane channel protein (tolC), and lipoprotein (nlpD_2).
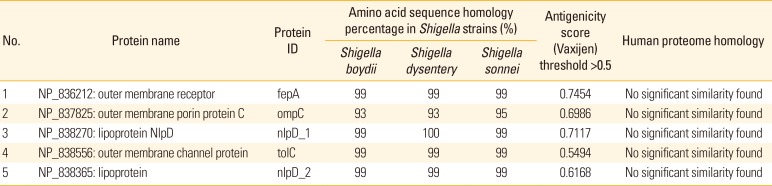
Table 1
Protein alignment and antigenicity prediction of the selected outer membrane proteins from Shigella flexneri 2a
![]()
Identification of human homologs
Potential vaccine candidates should be non-human homologous to avoid interference against host immune mechanism. Five OMPs selected from the above step were subjected to BLASTp analysis against human proteome. Five OMPs, notably outer membrane receptor (fepA), outer membrane porin protein C (ompC), lipoprotein NlpD (nlpD_1), outer membrane channel protein (tolC), and lipoprotein (nlpD_2) were observed to have no identity conserved with human proteome.
Protein physico-chemical properties analysis
Secreted and cell-surface proteins possess crucial role in inter-cellular communications. Secreted and a great number of cell surface proteins typically possess an N-terminal signal peptide which directs the proteins to the secretion apparatus. SignalP ver. 4.1 server was employed to predict the presence and location of signal peptide cleavage sites in amino acid sequences of each selected OMPs. Outer membrane receptor (fepA), outer membrane porin protein C (ompC), lipoprotein NlpD (nlpD_1), outer membrane channel protein (tolC), and lipoprotein (nlpD_2) were predicted consisting of N-terminal signal peptide cleavage sites between aa22–aa24. No signal peptide was observed in lipoprotein (nlpD_2). Physico-chemical properties analysis showed that the molecular weight and the theoretical isoelectric point of the five selected OMPs were ranging between 27.56 to 82.09 kDa and 4.56 to 10.21, respectively. Physico-chemical properties of five selected proteins are summarized in Table 2.
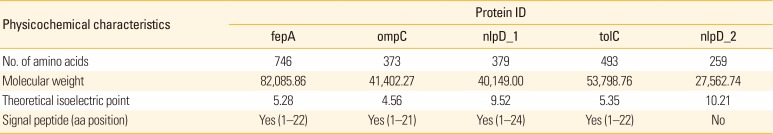
Table 2
Characterization of physico-chemical properties of vaccine candidates
![]()
Protein-protein interactions analysis
Interacting of a protein at cellular level could be helpful to have a better insight of the protein functional activities which could be of importance for potential therapeutic target. To explore the functional and interacting pathways of the five selected OMPs of S. flexneri 2a, STRING ver. 10.5 was used for protein networking analysis. As shown in Fig. 4, the topological analysis of the constructed protein network indicated a multiple nodes and edges interaction network map. Protein interacting network analysis revealed a total number of 15 nodes and 30 edges with 4.0 average node degree. fepA, ompC, nlpD_1, nlpD_2, and tolC were predicted to have involved in multiple interactions. fepA was observed to interact directly with fepB, fes, tonB, and ompC. OmpC showed direct interaction with proteins including with tolC, ton B, and rpoS. nlpD_1 and nlpD_2 could interact directly with rpoS but was observed to interact indirectly with ompC through rpoS. Despite having direct interaction with ton B and ompC, tolC was predicted to interact with emrK, acrA, acrB, macA_3, macB, and acrD. Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed that interaction of targeted OMPs of S. flexneri 2a selected in this study are essential for beta-lactam resistance pathway (pathway ID no., 00312), indicating diverse proteins jointly contribute to a shared function at cellular level.
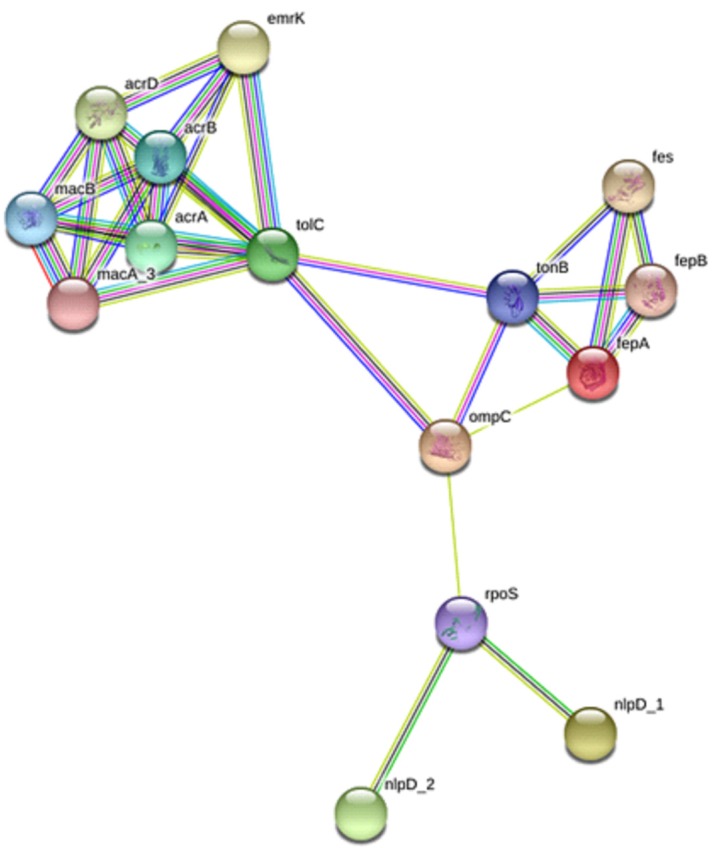 | Fig. 4Protein-protein interaction network of five outer membrane proteins (fepA, ompC, nlpD_1 and nlpD_2, and tolC) with other neighboring proteins in Shigella flexneri 2a. Input protein: fepA (outer membrane receptor), ompC (porin), nlpD_1 (lipoprotein NlpD), nlpD_2 (lipoprotein), and tolC (outer membrane channel protein). Predicted functional partners: acrA (multidrug efflux system transporter AcrA), acrB (multidrug efflux system protein AcrB), macB (macrolide transporter ATP-binding/permease), tonB (transporter), rpoS (RNA polymerase sigma factor RpoS), macA_3 (macrolide transporter subunit MacA), fepB (iron-enterobactin transporter periplasmic binding protein), fes (enterobactin/ferric enterobactin esterase), emrK (multidrug resistance protein K), and acrD (aminoglycoside/multidrug efflux system).
|
B-cell epitope prediction
For the prediction of surface-exposed B-cell epitopes in the five selected OMP of S. flexneri 2a, the full length amino acid sequences of each selected proteins were predicted using combination of two algorithms available in BCpreds server. These algorithms generated 20-mer sequences of B-cell epitopes with score >0.80. The combination of these algorithms allowed the program to predict a maximum of 54 B-cell epitopes from five OMPs with the length ranged from 20 to 41 amino acids. Each of these B-cell epitopes was then calculated for their antigenicity using Vaxijen ver. 2.0. Up to of five B-cell epitopes predicted respectively from ompC, nlpD_1, tolC, and nlpD_2 were eliminated from the list due to their low antigenic score <0.4. Number of B-cell epitopes present in five selected OMP of S. flexneri 2a is summarized in Table 3.
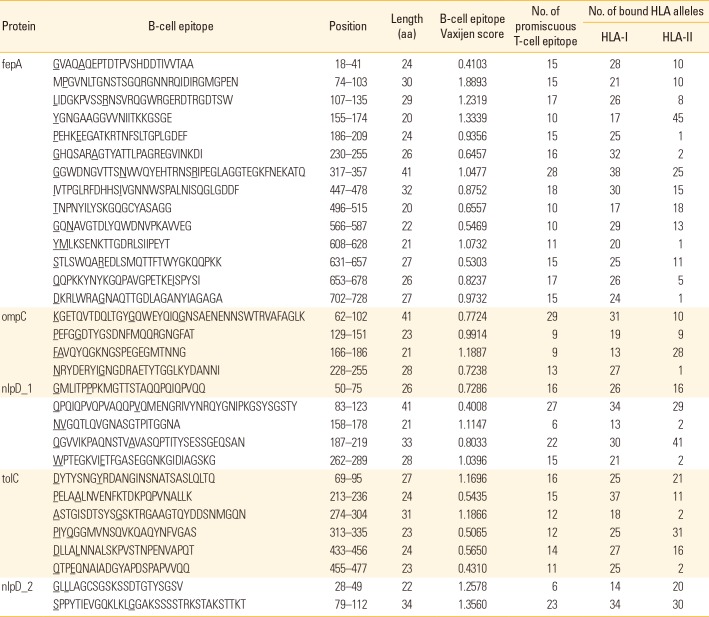
Table 3
Potential epitopes of five selected outer membrane proteins from Shigella flexneri 2a that can elicit both B-cell and T-cell immune response
![]()
T-cell epitope prediction
In order to identify promiscuous T-cell epitopes within the B-cell epitopic sequences, surface-exposed B-cell epitopes of each selected OMP were further evaluated for their effective binding to HLA class I and HLA class II alleles using Propred [24] and Propred I servers [25], respectively, based on quantitative matrices. A total of 31 antigenic regions of the five selected OMPs were predicted to contain both promiscuous B- and T-cell epitopes (Table 3). Of these, fourteen epitopes was predicted in fepA while four epitopes were predicted in ompC, six epitopes were in nlpD_1 and tolC, respectively, and two epitopes were in nlpD_2. All of these antigenic epitopes were predicted to be able to bind to at least 15 alleles corresponding to HLA class I and HLA class II. Among the highest number of binders, B-cell epitope corresponded to ompC (62KGETQVTDQLTGYGQWEYQIQGNSAENENNSWTRVAFAGLK102) was found to contain 29 promiscuous T-cell epitopes with the ability to bind to 31 HLA class I and 10 HLA class II, respectively; whereas 22 promiscuous T-cell epitopes lie within B-cell epitope of nlpD_1 (187QGVVIKPAQNSTVAVASQPTITYSESSGEQSAN219) were predicted to bind to 30 HLA class I and 41 HLA class II, respectively.
Go to :
Discussion
Diarrhoeal disease caused by shigellosis continues to be a leading cause of morbidity and mortality worldwide. Without a proper control, the infection could potentially lead to major human life-threatening disease. Increased resistance level in Shigella to a wide range of antibiotics has led the disease to a global health issue. Until now, efforts to develop safe and efficacious vaccines against Shigella are still unsuccessful. Attenuated live vaccines developed for human were found not to be safe partly due to their possibility in occasional back mutations. Later, lipopolysaccharide-based vaccines containing the sugar moiety were also reported to have low immunogenicity when evaluated in infants and young children [27]. The emergence of multiple drug resistance in Shigella and lack of vaccine against Shigella have necessitated a search for alternative strategies to further discover potential vaccine targets against Shigella infections.
The advent of genome sequencing and advancement in bioinformatics is now possible to identify potential vaccine candidates computationally without the need for living culture. This approach termed as “reverse vaccinology” reduces the time and cost of designing and identifying vaccine candidates, it also provide us with new solutions for those diseases for whom conventional approach have been unsuccessful [28]. The first vaccine developed using this approach was against the serogroup B Neisseria meningitidis [12]. In this study, we adopted “reverse vaccinology” method to identify potential OMPs from S. flexneri 2a that conserved among S. dysentery and S. sonnei for vaccine development. It is interesting to note that bacterial cell surface and secreted antigens could be important targets for developing vaccine given that they are responsible for the initial host-pathogen interaction [9]. Based on the proteins sequence conservedness, immunogenicity, subcellular localization, and unrelatedness with human proteome, five OMPs were identified as potential vaccine candidates against S. flexneri. The selected proteins were outer membrane receptor (fepA), outer membrane porin protein C (ompC), lipoprotein NlpD (nlpD_1), outer membrane channel protein (tolC), and lipoprotein (nlpD_2).
Surface proteins of many pathogens are mostly antigenic and are responsible for host pathogenesis [29]. During colonization, the attachment of pathogens to host cells surfaces is usually mediated by adhesins primarily built up of OMPs. OMPs, which are characterized as a large group of β-barrel proteins found in the outer membrane of Gram-negative bacteria have been reported to possess multiple roles during bacteria pathogenesis including mediating of the initial adhesion of bacteria to host cells, modulation of host-pathogen interactions as well as propagation of virulence factors [30]. It is well documented that OMPs of Gram-negative bacteria consist of components which are easily recognized as foreign antigens by immunological defense systems of the hosts [31]. This strongly suggests that OMPs of Gram-negative bacteria are essentially to play crucial roles both in the process of bacterial pathogenesis and stimulation of the host immune response. In view of this, OMPs of Shigella have been reported to confer a significant protective immune response [32]. ompA has been identified as a major protective antigen and antigenically conserved among the Shigella spp. Immunization of mice with ompA was found to give protection against a lethal dose of virulent S. flexneri 2a [10]. Meanwhile, previous study also showed that mice immunized with ompC survived after being challenged with live S. flexneri 3a [33], suggesting the OMPs could be one of the ideal components for developing subunit vaccine against shigellosis.
Despite having essential role in pathogenesis, the bacterial membranes also known to possess functions as defensive barriers to protect the cells from harmful and toxic compounds [34]. In order to understand the functional and cellular processes at system-level, protein-protein interacting network within the outer membrane of the bacteria is of importance. In this study, protein-protein interaction analysis revealed that five selected S. flexneri 2a OMPs were involved primarily in beta-lactam resistance pathway (pathway ID no., 00312). Beta-lactamases are enzymes produced by bacteria that provide multi-resistance to beta-lactam antibiotics. Bacterial resistance to antibiotics can be associated with the degradation of the drug by these enzymes [35]. It has been reported that resistance toward beta-lactam antibiotic is associated with alteration in outer membrane porins ompC and ompF and cytosolic proteins of ompR in Shigella [36]. Hence, active efflux pump is another major factor responsible for the antibiotic resistance phenotype in Gram-negative bacteria [37]. Previous studies have shown that AcrAB-TolC pump is involved in multidrug resistance in Gram-negative bacteria including Escherichia coli, Enterobacter aerogenes, Salmonella enterica, and S. flexneri [38]. The drug metabolism can be associated with overexpression of acrA gene which leads to multiple-antibiotic resistance in clinical isolates of S. flexneri [38]. In this study, two OMPs (fepA and tolC) identified from the selection pipeline are among the OMPs reported to be involved in facilitating the internalization of the external irons into the bacterial cell for bacterial colonization and survival [39]. The data presented here indicates that five OMPs of S. flexneria 2a selected in this study may possess essential role in facilitating drug resistance for bacterial survival, supporting these proteins could serve as potential vaccine targets against multidrug resistant Shigella spp.
Shigella spp. infection involves both intracellular and extracellular phases. During invasion, the bacteria target macrophages and gut colonic epithelial cells. To evade host immune system, Shigella uses its T3SS-dependent mechanism to avoid rapid clearance by promoting in vivo apoptosis of immune cells in mucosa-associated lymphoid tissues of the gut, resulting in innate immune response is insufficient to control Shigella spp. infection [40]. Following entering the epithelial cells, Shigella spp. can elicit both the humoral and cell-mediated immune responses which are responsible to facilitate the clearance of Shigella spp. infection from the host [41]. Given the crucial role of adaptive immunity in protection against Shigella spp. infection, the selection of antigens containing both B- and T-cell epitopes may provide an efficient way for vaccine development against Shigella spp. [41]. In this study, five OMPs selected by immunoinformatics method were found to possess numbers of antigenic epitopes which could potentially induce both humoral (B-) and cellular (T-cell) mediated immune responses [42]. The immunogenic epitopes predicted from each of these OMPs are potentially to be used for designing multi-epitope vaccines against this bacterium. Therefore, computationally or better known as reverse vaccinology approach is both cost effective and less time consuming for screening of potential vaccine candidates and further providing a framework for future designing of an effective vaccine against Shigellosis. The selected vaccine candidates need to be further validated for their immunogenicity and protective efficacy experimentally for their future use as vaccine targets against various strains of Shigella spp.
In summary, five target OMPs of S. flexneri 2a selected through our bioinformatics pipeline have been verified computationally as potential vaccine candidates against S. flexneri 2a. Our data shown that these OMPs are conserved among the Shigella strains with particular S. boydii, S. dysentery, and S. sonnei and therefore suggested they are potentially effective against all the strains of Shigella spp. Reverse vaccinology based on the data derived from genomics, proteomics, and immunoinformatics is a promising strategy for the screening and identification of antigenic antigens which could have potential capacity to evoke cellular and humoral immune responses simultaneously against Shigella spp. infection. Further in vitro and in vivo experiment studies are required to validate the immunogenicity and protective efficacy of these antigens against experimentally Shigella infection in animal model. The current user-friendly vaccine identification pipeline can be extended to other target pathogens to pave a better way for the control of transmission of persistent infections.
Go to :
 XML Download
XML Download