

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Cerebellar ataxia is characterized by impaired balance and coordination caused by cerebellar dysfunction [1]. In general, dysmetria, limb ataxia, dysdiadochokinesis, or intention tremor are observed during physical examination. Cerebellar ataxia has a heterogeneous etiology and manifests as either acute or chronic forms. The most common causes of acute cerebellar ataxia include vascular disorders such as ischemia or hemorrhage, toxins, and medications. Chronic progressive ataxia is the most common form of hereditary cerebellar ataxia, although its rate of progression and severity vary depending on the causative gene.
Hereditary cerebellar ataxia usually has an autosomal pattern of inheritance, including spinocerebellar ataxia (SCA), Friedreich's ataxia, and ataxia-telangiectasia. SCAs are autosomal dominant ataxias classified as SCA1 through SCA36 according to the genetic loci [2]. Friedreich's ataxia is an autosomal recessive disorder and ataxia-telangiectasia is associated with defective DNA repair [34]. Although it is uncommon, de novo mutation in various genes related to cerebellar function can trigger cerebellar ataxia.
Here, we present a 33-year-old male patient with cerebellar ataxia. He was initially considered to have a psychiatric conversion disorder without organic causes. However, a deletion in chromosome 7q31.2-31.32 including Ca2+-dependent activator protein for secretion (CADPS) gene was discovered by targeted gene sequencing using a next-generation sequencing (NGS) panel and chromosomal microarray. To the best of our knowledge, the role of this chromosomal deletion in 7q31.2-31.32 involving CADPS gene in a patient with cerebellar ataxia has never been reported.
CASE DESCRIPTION
A 33-year-old male visited the clinic of our hospital with complaints of weakness in bilateral lower extremities and balance problem. At first, he could walk independently until 2016 and perform daily living activities without assistance. For no particular reason, he recognized weaknesses in both lower extremities since January 2017 with slowly worsening symptoms. His symptoms showed chronic and progressive patterns. Several examinations including brain magnetic resonance imaging (MRI) and C-spine MRI performed at the Department of Neurology in other hospitals revealed no specific findings. When he was referred to our hospital in April 2017, he showed weakness in both legs along with poor standing balance, resulting in gait disturbance without hand support.
He was born without any complications. There was no notable family history. Although he started to walk at 20 months, he grew up within normal range. During his school days, he showed poor academic performance and was bullied by his class-mates. He had difficulties with getting along with his colleagues in the workplace. He has been on medication for depression and insomnia since October 2016.
Due to his psychiatric history and relatively mild neurological symptoms, the possibility of malingering or other psychiatric diseases was suspected. Thus, a psychological interview was conducted at other hospital to assess the relevance of motor weakness and psychological causes. The interview revealed significant distress associated with relationships with family members and colleagues. Thus, a diagnosis of conversion disorder was suggested.
Manual muscle test revealed grade 4 in bilateral lower extremities. No sensory impairment was detected. The patient presented with no signs of spasticity involving all four extremities. Dysmetria was marked in finger-to-nose and heel-to-shin test. A clumsy response was found in rapid alternating movement evaluation. Romberg sign and tandem gait were undetectable due to poor standing balance. The patient scored 26 points on the Berg balance scale. His activities of daily living were not independent because of weakness and balance problem. His modified Barthel index score was 78 points at initial evaluation (Table 1).
Table 1
Changes in Berg balance scale and modified Barthel index in a patient with cerebellar ataxia

| Initial | 6 months | 12 months | 18 months | |
|---|---|---|---|---|
| Berg balance scale | 26 | 40 | 46 | 54 |
| Modified Barthel index | 78 | 86 | 92 | 91 |
![]()
To determine the causes of his symptoms, we performed electrophysiological studies to evaluate nerve conduction and somatosensory evoked potentials. Results did not show any evidence of peripheral neuropathy or central conduction defect. Laboratory results for anti-GM1 antibody, anti-GD1b antibody, and anti-myelin-associated glycoprotein antibody were all negative. Official findings of brain MRI and C-spine MRI were within normal limits. Since cerebellar ataxia was found in physical examinations, we decided to perform brain positron emission tomography-computed tomography (PET-CT) scan despite normal brain MRI findings, which revealed a decreased fluorodeoxyglucose (FDG) uptake in the anterior cerebellum (Fig. 1).
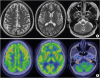 | Fig. 1Brain MRI and PET-CT images in a patient with cerebellar ataxia.There was no remarkable finding in brain MRI images (A), although hypometabolism in the anterior cerebellum was noted (B, arrow).
MRI, magnetic resonance imaging; PET-CT, positron emission tomography-computed tomography.
|
Because there was no vascular insult or trauma history to explain the patient's cerebellar dysfunction, we also performed targeted gene sequencing by using a NGS panel including 172 target genes associated with neuropathy. Massively parallel sequencing was done using the MiSeq System (Illumina, Inc., San Diego, CA, USA). Quality control and sequence analysis were performed with the BaseSpace (Illumina, Inc.) and NextGENE (SoftGenetics, State College, PA, USA) software programs and cross-validated with our custom analysis pipeline. Although whole-exome sequencing was not used for analysis, targeted NGS panel showed chromosomal deletion at 7q31.2-31.32 and possible copy-number variation (Fig. 2).
 | Fig. 2Next-generation sequencing analysis in a patient with cerebellar ataxia.There was a suspected finding of chromosomal deletion in 7q31.2-31.32.
|
To confirm the chromosomal deletion, we further conducted chromosomal microarray analysis and whole-chromosome fluorescence in situ hybridization (FISH). The FISH results using whole-chromosome probes showed a 7q deletion. Comparative genomic hybridization was performed with oligonucleotides (microarray) using the Affymetrix Cytoscan 750K and found a deletion of approximately 8.6 Mb in the ‘q’ arm of chromosome 7 involving 7q31.2-31.32 band (Fig. 3). Among the affected genes involving 7q31.2-31.32 band in the OMIM database, CADPS2 is known to be associated with autism spectrum disorder and cerebellar development [56].
 | Fig. 3Chromosomal microarray analysis in a patient with cerebellar ataxia.There was a deletion of approximately 8.6 Mb in the ‘q’ arm of chromosome 7 involving 7q31.2-31.32 band.
|
The patient was provided with balance training and progressive gait training. After 12 months, Berg balance scale was improved from 26 to 46 and modified Barthel index was increased from 78 to 92 (Table 1). However, he could not walk independently without using an anterior walker at the time. We prescribed a trial with dopamine agonist, pramipexole, to determine its effectiveness for ataxia and trunk control. Six months after treatment with a dopamine agonist, the patient is able to walk without the need for a walker. The patient's Berg balance scale was improved again from 46 to 54 (Table 1).
DISCUSSION
The chief complaint of the patient was weakness of both legs. However, no specific lesion was detected using brain MRI or C-spine MRI. Cerebellar ataxia was detected during physical examination and cerebellar FDG uptake decreased in brain PET-CT scan. The related genetic abnormality was then confirmed. In the absence of relevant evidence based on conventional tests, it may be necessary to consider genetic studies.
CADPS family is known to regulate secretion of neuropeptides including brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) from dense-core vesicles [7]. The two members of CADPS family include CADPS1 and CADPS2. CADPS1 is located in 3p14.2 and Speidel et al. [8] reported that CADPS1 is necessary for catecholamine loading in large dense-core vesicles. On the other hand, CADPS2 is located in 7q31.32 and it is found in the cerebellum. It plays a crucial role in cerebellar development by releasing BDNF and NT-3. Sadakata et al. [5] have reported that CADPS2-deficient mice show suppressed phosphorylation of Trk receptors in the cerebellum and impaired neuronal survival, lobulation, and synaptogenesis during cerebellar development. Thus, it is possible that CADPS2 deletion is associated with symptoms of cerebellar ataxia in this patient, considering the location of the gene mutation.
CADPS2 variants are known to be associated with autism spectrum disorders. Sadakata et al. [9] have reported that CADPS2-knockout mice show autistic-like behaviors. Aberrant CADPS2 mRNA has been found in autistic patients. Grabowski el al. [6] have reported a patient diagnosed with autism spectrum disorders associated with recurrent psychotic syndrome and a deletion involving the 7q31-32 band at the CADPS2 gene locus.
Cerebellar ataxia is not directly related to autistic disorders. In a previous study, however, some of the autistic patients were found to manifest cerebellar abnormalities [10]. Although autism was not clearly diagnosed in this patient, it might be included in the spectrum of autistic behavior disorders based on Diagnostic and Statistical Manual of Mental Disorders, 5th Edition, considering his challenges with social life, including school and workplace.
This patient showed a significant functional improvement based on Berg balance scale and modified Barthel index after intensive and repetitive balance training with psychological support. He received medications including ginkgo biloba extract, norepinephrine-dopamine reuptake inhibitor (bupropion), and beta-blocker (propranolol). However, the medications were not effective to control his symptoms. Based on a report that autism symptoms were improved by administrating dopamine agonist in the CADPS2 knock-out mouse model [9], a dopamine agonist (pramipexole) was tried to control ataxic symptoms. After taking a dopamine agonist for 6 months, he could walk independently without using an anterior walker. A further study is needed to determine if dopamine agonist represents an alternative treatment of choice for cerebellar ataxia with CADPS gene deletion.
In conclusion, we present a 33-year-old male with cerebellar ataxia who was found to have chromosomal deletion in 7q31.2-31.32 involving CADPS gene. Considering that there is no reported case of cerebellar ataxia associated with CADPS2, it is important to consider various gene mutations as the possible etiological factors for ataxia after excluding the common causes.
 XML Download
XML Download