

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Kawasaki disease (KD) is an acute, self-limiting febrile illness that is characterized by systemic inflammation involving all medium-sized arteries and multiple organs during the acute febrile phase, leading to associated clinical findings [1]. The diagnosis of KD is based on the presence of principal clinical findings: fever, extremity changes, rash, conjunctivitis, oral changes, and cervical lymphadenopathy [1]. However, the diagnosis of KD is sometimes overlooked or delayed because other systemic organ manifestations may precede such principal clinical features of KD, especially in early infant and older children [2]. Common clinical scenarios mistakenly diagnosed before the diagnosis of KD are urinary tract infection (pyuria), aseptic meningitis (irritability and a culture-negative pleocytosis of the cerebrospinal fluid), bacterial adenitis (retropharyngeal phlegmon), and bacterial sepsis (septicemic shock) [3]. Cardiovascular manifestations may include myocarditis, pericarditis, valvular regurgitation, shock, and coronary artery abnormalities [3]. Regarding pericarditis, echocardiographic findings of a small pericardial effusion are common, but pericardial effusion large enough to manifest clinical signs of cardiac tamponade is very rare [45]. Here, we describe a case of incomplete KD presenting with impending cardiac tamponade and hemorrhagic pleural effusion.
CASE REPORT
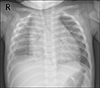
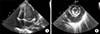
A previously healthy 10-month-old girl visited the emergency room due to high fever for 6 days, diarrhea, and respiratory distress. The past medical history was unremarkable. On arrival, the patient looked acutely ill and showed tachypnea with chest wall retraction. The blood pressure was 72/52 mmHg, heart rate 167/min, respiratory rate 43 breaths/min, and the oxygen saturation 86%~88%. Initial laboratory findings showed white blood cell (WBC) count of 33.6×109/L (neutrophils 84.5%, lymphocytes 13%), erythrocyte sedimentation rate (ESR) of 43 mm/h (normal range <20), and C-reactive protein of 31.47 mg/dL (normal range <1.0). The blood urea nitrogen and creatinine were 65 mg/dL and 1.02 mg/dL, respectively. Serum pro-brain natriuretic peptide level (BNP) was 13,835 pg/mL. Urine analysis showed pyuria (20~29 WBCs per high-power field and negative nitrate test). Electrocardiogram showed ST elevation in leads V4–6. Chest radiography showed massive cardiomegaly with pleural effusion in the right thoracic cavity (Figure 1). Echocardiography revealed a large amount of pericardial effusion with impending cardiac tamponade, but normal ventricular systolic function and coronary arteries without giant aneurysm and aneurysmal rupture (Figure 2). Under the impression of acute pericarditis with impending cardiac tamponade and sepsis, an emergency pericardiocentesis was performed and serosanguinous fluid was drained. The results of pericardial fluid analysis were as follows: red blood cell count 300×106/L, WBC count 570×109/L with 85% of neutrophils, lactate dehydrogenase (LDH) 26,900 IU/L (serum LDH 1,245 IU/L), and protein 3.87 g/dL (serum protein 5.1 g/dL). Exudative pericardial effusion was diagnosed based on pericardial fluid to serum protein ratio >0.5 and pericardial fluid to serum LDH > 0.6 (Light's criteria). In addition, the fluid drained through thoracentesis was hemorrhagic (red blood cell count 9,920×106/L, the ratio of pleural fluid to serum LDH >0.6). Empirical antibiotics (cefotaxime and vancomycin) were administrated intravenously. Culture studies from blood, urine, and pericardial and pleural fluid failed to isolate any pathogen. Despite a regimen of multiple empirical antibiotics, high fever persisted. On the 19th day of fever, conjunctival injection and desquamation of both fingertips suggesting an incomplete KD developed. Follow-up laboratory tests were performed and met the criteria of supplemental laboratory findings supporting a diagnosis of incomplete KD (elevated ESR [25 mm/h] and CRP [20.73 mg/dL], low serum albumin [2.7 g/dL, reference range: 3.5~5.2], anemia for age [9.2 g/dL], WBC count [38.84×109/L], platelet count [576×109/L] and sterile pyuria) [3]. Peripheral blood cell morphology showed that leukocytosis, neutrophilia, and normocytic normochromic anemia. There was an insufficient feature of macrophage activation syndrome (MAS) or systemic onset-juvenile idiopathic arthritis (SoJIA) in clinical course and follow-up laboratory test: Triglyceride (165 mg/dL, reference range: 40~200), ferritin (210 ng/mL, reference range: 26.1~287.6), fibrinogen (243 mg/dL, reference range: 160~350) (Table 1). Follow-up echocardiogram also showed a mild dilatation of the right coronary artery (2.4 mm in diameter; Z score=2.7) without pericardial effusion. Intravenous immunoglobulin (IVIG, 2 g/kg) and oral high-dose aspirin (50 mg/kg/day) were administered. Fever subsided on the 2nd day of IVIG treatment. The patient was discharged 4 days after IVIG treatment (Figure 3). Three days after the discharge, she was readmitted with a 24-hour history of high fever and pleural effusion. Despite the second dose of IVIG (2 g/kg), fever persisted (body temperature >38.5℃). Fever and pleural effusion were resolved after treatment with high dose methylprednisolone (Figure 3). After discharge, high fever and pleural effusion redeveloped twice, on the 41st and 57th days of illness, and laboratory findings also showed leukocytosis (33.6×109/L of WBC count) and elevated ESR (43 mm/h) and CRP (34.1 mg/dL) on each occasion at the time of admission. After administration of infliximab (5 mg/kg), fever and pleural effusion subsided each time. After discharge following the 4th admission, the patient is doing well. Echocardiography performed 1 year after the illness showed normal coronary arteries and ventricular function.
DISCUSSION
KD is a systemic vasculitis that can affect various organs [1]. The diagnosis of classic KD is based on the presence of ≥5 of 6 principal clinical criteria. Patients who have insufficient principal clinical features (fever ≥5 days and 2 or 3 compatible clinical criteria) may be diagnosed with incomplete KD, based on supportive laboratory findings or a positive echocardiogram [3]. However, KD diagnosis is sometimes delayed because other systemic manifestations may precede such principal clinical features of KD. There are various unusual initial presentations that delay diagnosis of KD, such as acute surgical abdomen [6], aseptic meningitis [7], cervical adenitis [8], and shock syndrome [9]. Zulian et al. [6] reported 10 cases of KD with acute surgical abdomen as an initial presentation, out of which 7 patients were diagnosed after surgical intervention. A prolonged fever and a culture-negative pleocytosis of cerebrospinal fluid suggestive of aseptic meningitis in a neonate may lead to an oversight of the diagnosis of KD, because enteroviral meningitis is common in this age group [7]. Kanegaye et al. [8] reported that cervical adenitis and fever were initial presentations in 57 KD patients and suggested that incomplete KD should be considered in patients with cervical adenitis unresponsive to empirical antibiotics. In this case, incomplete KD was suspected and diagnosed only after development of conjunctivitis and desquamation on the fingertips on the 13th day of hospitalization (the 19th day of fever).
Because clinical criteria are used to diagnosis of KD, patients who have insufficient principal clinical features tend to make early diagnosis difficult [3]. Moreover KD, SoJIA and MAS have similarity that clinical features and laboratory findings tend to overlap [10]. Therefore, the differential diagnosis of incomplete KD presenting especially unusual manifestation has diagnostic dilemma and clinical challenging, which often can be delay in diagnosis [210]. In our case, we tried to rule out infectious disease, drug-related polyserositis and connective tissue disease. According to the laboratory finding including bacterial cultures and virus studies, infectious disease could be excluded. The absence of signs of arthritis during 1 year follow-up, negative study of auto-immune antibodies (anti-nuclear antibody, rheumatoid factor), and other laboratory features (ferritin, triglyceride, fibrinogen, WBC count, platelet, etc.) were insufficient to diagnose of connective tissue disease especially SoJIA and MAS [1011].
KD with massive pericardial effusion showing clinical signs of cardiac tamponade is very rare. There have been several reports on hemorrhagic pleural and/or pericardial effusion in patients with KD [451213]. Voynow et al. [13] reported that 11 (1.83%) out of 602 KD patients presented with predominant pulmonary involvement. However, pericardial involvement is usually manifested as a small amount of pericardial effusion on the echocardiogram, and to our knowledge, only 2 cases with cardiac tamponade have been reported [45]. Our case showed an atypical course where massive pleural effusion and pericardial effusion with cardiac tamponade without giant aneurysm and aneurysmal rupture developed as initial presentations in an infant under 12 months.
Currently, first-line treatment of KD is a single dose of IVIG (2 g/kg) and second-line treatment for IVIG-resistant KD patients may be a repeat dose of IVIG, high-dose methylprednisolone, or infliximab [3]. It is known that the levels of pro-inflammatory cytokines (interleukin [IL]-2, IL-6, IL-8, interferon-γ and tumor necrosis factor [TNF]-α) and vascular endothelial growth factor (VEGF) are increased in KD [1415]. Therefore, infliximab (anti-TNF-α monoclonal antibody) therapy is preferable to use in patients with KD refractory to IVIG [3]. Other monoclonal antibody therapy (anakinra, a recombinant, nonglycosylated form of the human IL-1 receptor antagonist ) for treatment of refractory KD was reported [16]. Regarding the pathogenesis of pleural and pericardial effusion in KD, it has been speculated that VEGF may play a role by increasing microvascular hyperpermeability [12]. Strunk et al. [17] reported that infliximab and glucocorticoids reduced VEGF in patients with rheumatoid arthritis. However, a study in KD showed that infliximab reduced pro-inflammatory cytokines but did not suppress VEGF in refractory KD patients [14]. Instead, Hamada et al. [12] reported a case that presented with marked pericardial effusion and pleural effusion with elevated VEGF, which were dramatically resolved after corticosteroid administration. In our case, recurrent fever and pleural effusion developed even during steroid administration, and showed acute response to infliximab therapy, suggesting its partial role in reducing the hyperpermeable state of KD. Further study is needed to elucidate the possible roles and mechanisms of infliximab and corticosteroid in suppressing vascular hyperpermeability in KD patients.
SUMMARY
KD is included on the list of important causes of MAS in childhood, and the large degree of overlap syndrome should be considered the possibility of similar relationships between refractory incomplete KD and MAS [18]. It is important to remember that in infants with unexplained persistent fever and irresponsive to empirical antibiotics, incomplete KD should be suspected. In addition, clinicians should be aware that cardiac tamponade and/or hemorrhagic pleural effusion may be initial clinical manifestations of incomplete KD.


 XML Download
XML Download