

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Systemic lupus erythematosus (SLE) is characterized by immune regulation disruption and multisystem involvement and is mediated by autoantibodies and immune complex deposits [1]. Disrupted immune regulation via dysregulation of B- and T-cell activation and aberrant production of cytokines plays a key role in SLE pathogenesis [2]. Although the etiology of SLE is incompletely understood, it is clear that genetic components play key roles in SLE pathogenesis [23].
A subtype of T cells, Th17 cells, secrete interleukin-17 (IL-17), which is a pleiotropic pro-inflammatory cytokine that enhances T-cell priming and stimulates epithelial, endothelial, and fibroblastic cells to produce multiple proinflammatory mediators such as tumor necrosis factor-alpha (TNF-α), IL-1β, IL-6, and chemokines [4]. IL-17 plays a critical role in innate and adaptive immune systems by promoting inflammation, cytokine production, B-cell proliferation, and autoantibodies production [4]. IL-17 consists of six protein members [IL-17A, IL-17B, IL17-C, IL-17D, IL-17E (IL-25), and IL-17F] of which IL-17A and IL-17F are responsible for the activity of Th17 cells in the induction of other cytokines and chemokines [5]. IL17A and IL17F are both located on chromosome 6p12, which is a genomic region linked to SLE [6]. In addition to the ability of IL-17 to regulate inflammatory reactions and immune responses and because of their chromosomal location, these genes are considered potential candidate genes for SLE.
Several studies investigating circulating IL-17 levels in patients with SLE compared to healthy controls and testing polymorphisms in different IL17-encoding genes have found associations with SLE susceptibility [78910111213141516171819]. Thus, we performed a meta-analysis to overcome the limitations of individual studies and to resolve the inconsistencies in their findings. The aim of our meta-analysis was to review systematically available evidence on serum/plasma IL-17 levels in patients with SLE compared to those found in controls and to determine whether polymorphisms in IL17-encoding genes were associated with SLE susceptibility.
MATERIALS AND METHODS
Identification of eligible studies and data extraction
We performed a literature search for studies that examined IL-17 levels in patients with SLE and controls, evaluated the relationship between circulating (serum or plasma) IL-17 levels, or tested for associations between polymorphisms in IL17 genes and SLE. PUBMED, EMBASE, and Cochrane databases were searched to identify all available articles (up to August 2019). The following key words and terms were used in the search: “IL-17”, “level OR serum OR plasma”, “polymorphism”, “systemic lupus erythematosus”, and “SLE”. In addition, all references cited were reviewed to identify additional studies that were not included in the above-mentioned electronic databases. Studies were considered eligible based on the following inclusion criteria: (1) they were case-control, cohort, or cross-sectional studies; (2) they provided data on IL-17 levels in both affected and control groups; (3) they included at least 10 patients with SLE; and (4) they tested IL17 gene polymorphisms in SLE and control groups. Studies were excluded if: (1) they contained overlapping or insufficient data or (2) they were reviews or case reports. Data on the methods and results were extracted from original studies by two independent reviewers. Discrepancies between the reviewers were resolved by consensus. We performed the meta-analysis in accordance with PRISMA guidelines [20]. The following information was extracted from each study: primary author, year of publication, country, ethnicity, adjustments for age and sex, number of participants, mean and standard deviation (SD) of IL-17 levels, and allele and genotype frequencies of polymorphisms in IL17 genes. When data were presented as medians, interquartile ranges, or ranges, the mean and SD values were derived using previously described formulae [2122].
Evaluation of statistical associations
We performed a meta-analysis to examine the relationship between IL-17 levels and SLE and to evaluate the allelic effect of the minor allele versus the major allele of different polymorphisms in IL17 genes. For continuity of data, results were presented as standardized mean differences (SMDs) and 95% confidence intervals (CIs). Odd ratios (ORs) and 95% CIs were calculated for dichotomous data. We assessed within-study and between-study variations and heterogeneities using Cochran's Q-statistics [23]. The heterogeneity test was used to assess the null hypothesis that all studies were evaluating the same effect. When the Q-statistic was significant (p<0.10), indicating heterogeneity across studies, a random effect model was used for the meta-analysis; otherwise, a fixed effect model was applied [24]. It was assumed that all studies estimated the same underlying effect and specifically considered within-study variation [23]. We quantified the effect of heterogeneity using I2=100% × (Q−df)/Q [25], where I2 was a measure of the degree of inconsistency between studies and determines whether the percentage total variation across studies was due to heterogeneity and not chance. I2 ranged from 0% and 100%. I2 values of 25%, 50%, and 75% were referred to as low, moderate, and high estimates, respectively [25]. Statistical determinations were performed using the Comprehensive Meta-Analysis computer program (Biosta, Englewood, NJ, USA).
Evaluation of heterogeneity, sensitivity test, and publication bias
To examine potential sources of heterogeneity observed in the meta-analysis, a meta-regression analysis was performed using the following variables: ethnicity, adjustment for age and/or sex, publication year, sample size, and data type. A sensitivity test to assess the influence of each individual study on the pooled effect size was performed by omitting each study individually. Although funnel plots are often used to detect publication bias, they require diverse study types of varying sample sizes, and their interpretation involves subjective judgment. Therefore, we assessed publication bias using Egger's linear regression test [26], which measures funnel plot asymmetry using a natural logarithm scale of the effect size.
RESULTS
Studies included in the meta-analysis
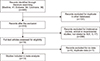
We identified 449 studies using electronic and manual search methods of which 19 were selected for full-text review based on the title and abstract, and six were excluded because they either lacked data or provided duplicated data. Therefore, 13 articles met our inclusion criteria [78910111213141516171819] (Figure 1). One of the eligible studies contained data on two different groups [19], which were treated independently. Fourteen comparisons were examined in the meta-analysis, which consisted of 2,096 patients with SLE and 2,587 controls (Table 1). Eight studies examined IL-17 levels in affected and control groups, and six comparative studies from five articles evaluated polymorphisms in IL17 genes in both the SLE and control groups (Table 1). Meta-analysis of IL17-related polymorphisms was performed if there were at least two comparisons. Because of the limited number of candidate-gene association studies, four types of meta-analyses were performed: rs2275913 in IL17A and rs763780, rs2397084, and copy number variations (CNVs) in IL17F. Characteristic features of the studies included in the meta-analysis are summarized in Table 1.
Meta-analysis of circulating IL-17 levels in patients with SLE compared to those in controls
Using meta-analysis, we found that IL-17 levels were significantly higher in the SLE group than those in the control group (SMD=1.045, 95% CI=0.521~1.568, p<0.001) (Table 2, Figure 2). Stratification based on ethnicity revealed higher IL-17 levels in the SLE group among Asian and Latin American populations but not in Arabs (Table 2). Subgroup analysis using sample size showed significantly higher IL-17 levels for large (n>100) but not small (n<90) sample groups in the SLE group compared to those found in the control group (Table 2).
Meta-analysis of IL17A rs2275913, IL17F rs763780, and IL17F rs2397084 polymorphisms and IL17F CNVs and SLE susceptibility
Our meta-analysis revealed no evidence of an association between the IL17A rs2275913, IL17F rs763780, and IL17F rs2397084 polymorphisms and SLE susceptibility (Table 3, Figure 3). However, our meta-analysis showed a significant association between SLE and IL17F CNVs in a pooled cohort of affected individuals compared to those found in pooled controls (OR=3.663, 95% CI=2.466~5.221, p<0.001) (Table 3, Figure 3).
Heterogeneity and publication bias
Between-study heterogeneity was identified during the meta-analyses of IL-17 levels in patients with SLE (Table 2). However, meta-regression analysis showed that ethnicity, data type, sample size, and adjustment for age and/or sex had no effect on heterogeneity in our meta-analysis of IL-17 levels in patients with SLE (all p>0.05). Sensitivity analysis showed that no individual study significantly affected the pooled SMD, indicating that the results of this meta-analysis were robust. No heterogeneity was found in the meta-analyses of polymorphisms in IL17 genes, with the exception of rs22759136 in IL17A and rs763780 in IL17F. However, the studies included in the meta-analysis for rs763780 showed the same directionality of the ORs (Figure 2). Publication bias results from a disproportionate number of positive studies and poses a problem for meta-analyses. However, we found no evidence of publication bias for any of the study subjects (i.e., the funnel plot showed no evidence of asymmetry, and from Egger's regression test all p>0.05).
DISCUSSION
In this meta-analysis, evidence for elevated circulating IL-17 levels in SLE and for association between polymorphisms in IL17 genes and SLE susceptibility were evaluated. Our meta-analysis of 13 studies, which included 2,096 patients with SLE and 2,587 controls, showed that circulating IL-17 levels were significantly higher in the SLE group than the control group. The reason of significantly higher IL-17 levels for large but not small sample groups is unclear, but it may be explained by statistical power due to sample size. In addition, we showed a significant association between IL17F CNVs and SLE susceptibility. These findings indicate that an increased IL-17 level may play a role in the pathogenesis of SLE and that CNVs from IL17-encoding genes may be associated with SLE risk. Our meta-analysis provides evidence that higher IL-17 levels are correlated with pathogenesis of SLE and, therefore, supports the involvement of Th17 cells in SLE. Th17 cells have been increasingly recognized as important mediators of autoimmune diseases [27]. Th17 cells are associated with the production of mediators of inflammation; thus, they have potentially pathogenic roles in autoimmune disease [27]. IL-17, a major effect factor of Th17 cells, is a potent pro-inflammatory cytokine that is produced by activated T lymphocytes and is also a potent inducer of TNF-α, IL-1-β, IL-6, IL-8, and G-CSF [4]. Our findings that IL-17 levels increase in the patients with SLE support a role of IL-17 in the pathophysiology of SLE.
Given the potential link between IL-17 and autoimmune diseases, polymorphisms in IL17 genes, which can affect IL-17 expression, have been studied as potential causes of autoimmune diseases [2829]. Our meta-analysis revealed an association between IL17F CNVs and SLE, in agreement with the results of functional studies conducted on the IL17F CNV polymorphism [19]. Increased copy numbers of IL-17F correlated with elevated mRNA levels [19]. Based on these findings and on those showing that IL17F CNVs are associated with both altered expression of IL-17, which can lead to elevated IL-17 levels, we propose a link between these variants and the development of SLE. However, our results on the association between the IL17A rs2275913, IL17F rs763780, and IL17F rs2397084 polymorphisms and SLE risk are not consistent with the functional studies of the polymorphisms. This may be because of the difference in clinical characteristics of the studied populations. In addition, genetic association results sometimes do not coincide with the results of functional studies for complex autoimmune diseases such as SLE. Multiple genes, genetic backgrounds, and environmental factors contribute to SLE development. Our negative results for the IL-17 polymorphisms might also be due to Type II error.
This meta-analysis has a few limitations. First, most of the recruited studies had small sample sizes, and a limited number of studies tested for evidence of an association between different polymorphisms in IL17 genes and its levels and SLE. Therefore, our meta-analysis may be underpowered. Second, the studies examined were heterogeneous with regard to both demographic characteristics and clinical features. Heterogeneity, confounding factors, and limited clinical information in these study populations may confound the results. Third, publication bias may adversely affect our analysis because studies with negative findings may not be published or identified in our search. Although we used Egger's regression test, the possibility of bias cannot be eliminated. Fourth, inflammatory cytokine level such as IL-17 varies according to disease activity, pre and post-treatment, and treatment drug types, even if measured in the same person. However, it is not possible to verify that all IL-17 levels of SLE patients were measured under the same conditions because individual papers do not indicate that they were all measured under the same conditions. Nevertheless, this meta-analysis also has its strengths. To the best of our knowledge, our meta-analysis is the first study that provides two parallel lines of evidence examining both IL-17 levels and polymorphisms in IL17-encoding genes in patients with SLE. While individual studies had a limited cohort size ranging from 24 to 576 participants, our pooled analysis had 2,096 patients. In addition, compared to individual studies, our study was able to provide data that were accurate by increasing the statistical power and resolution of the analysis through pooling the results of these independent analyses.
CONCLUSION
In conclusion, our meta-analysis demonstrated that circulating IL-17 levels were significantly higher in patients with SLE than the controls and that IL17F CNVs were associated with SLE susceptibility. Based on these findings, we conclude that IL-17 may have an important role in the pathogenesis of SLE. However, further studies are warranted to determine whether IL-17 levels directly contribute to the development of SLE.





 XML Download
XML Download