

 PDF
PDF ePub
ePub Citation
Citation Print
Print



I. Introduction
Drug–drug interaction (DDI) can be very harmful for patients and may cause serious health problems. When two or more drugs that can interact are administered, the patient is exposed to a potential DDI. DDIs cause up to 30% of adverse drug effects (ADEs). Thus, prevention of DDI plays a vital role in the treatment of patients. Moreover, adverse drug events are one of the primary reasons that drugs fail in clinical trials [1]. Besides their negative impact on public health, DDIs also result in significant economic losses of public resources. Prescribed interacting drugs cost tax payers billions of dollars. The number of new drugs entering the market is increasing every year (51 novel drugs were approved by the Food and Drug Administration in 2018) [2]. Thus, accurate prediction of potential DDIs is becoming even more crucial for the prevention of drug related harm. DDIs have been taken into consideration seriously by researchers. Many in-silico [3], in-vitro [4], and in-vivo [5] experiments have been carried out. However, since in-vitro and in-vivo experiments are extremely expensive and not always feasible, various in-silico research methods that provide successful results have been developed.
Ferdousi et al. [6] presented an approach, in which functional similarities of drugs in terms of protein targets are used for DDI prediction. Tatonetti et al. [7] created an adaptive data-driven approach to predict drug effects and interactions. They utilized a drug adverse effect database by correcting omissions of confounding factors, such as concomitant medications, patient demographics, and patient medical histories. Vilar et al. [8] proposed a method for the prediction of DDIs by utilizing known DDIs. They generated interaction profile fingerprint (IPF) vectors to represent drugs and used these vectors to calculate predictions. From a different perspective, Zhang et al. [9] utilized recommender methods to predict the unknown side effects of drugs. They used the integrated neighborhood-based method and restricted Boltzmann machine-based method as recommender methods. Vilar et al. [1] proposed a comprehensive method for creating drug fingerprints and DDI predictions. They created these fingerprints for known interaction profiles, adverse effects, and protein targets, as well as 2D and 3D molecular structures.
Ayvaz et al. [10] created a complete set of DDIs to obtain a comprehensive DDI list using publicly available DDI resources, such as DrugBank, KEGG, and the NDF-RT. Noor et al. [11] developed a novel pharmacovigilance inferential framework to infer mechanistic explanations for asserted DDIs and deduce potential DDIs. In another research study, Vilar et al. [1213] utilized molecular structures, generated fingerprints, and performed DDI predictions based on molecular similarities.
Similarly, the prediction of adverse drug reaction (ADR) has also been investigated extensively. Liu et al. [14] proposed a machine learning-based approach for ADR prediction by integrating the phenotypic characteristics of a drug, including indications, other known ADRs, the drug's chemical structures and biological properties, protein targets, and pathway information. In some studies, adverse event reports have been used to predict DDIs [151617]. Hudelson et al. [18] considered drug metabolism for DDI prediction. Percha et al. [19] applied a text mining algorithm to medicine abstracts to predict DDI. A text mining approach was also used by Tari et al. [20].
In this study, we evaluated and compared two of these approaches mentioned above [16]. These approaches were selected based on their DDI prediction accuracy and effectiveness. The first approach [1], further detailed in the following, gives very accurate prediction results among the described methods, whereas the second approach [6] considers similarities between known interaction pairs. Similarity calculations based on the second approach are simpler and computationally faster. Also, the similarity measure used in this approach provides more accurate results. Taking these points into consideration, these two approaches were evaluated. The first approach predicts DDIs using profile fingerprints depending on various attributes, such as interaction profiles [18], adverse effect profiles, and protein profiles [1]. In this approach, drugs are represented by vector fingerprints and known predictions. The similarities of all drug pairs are used to calculate the prediction matrix. On the other hand, the protein similarities between DDI drugs are calculated and used for the prediction of DDIs in the second approach [6]. The first approach achieves superior prediction accuracy in comparison to the second one. In addition, the DDI predictive power of interaction profiles, adverse effect profiles, and protein profiles were further evaluated.
II. Methods
1. Data Collection
In the study, the drug resources Merged-PDDI dataset [10] and MedDRA [21] were used. The dataset contained the DDIs from the Merged-PDDI-dataset, corresponding protein identifiers (carrier, transport, enzyme, target) from DrugBank [22] for those DDIs, and the adverse effects of the interacting drugs were retrieved from MedDRA. Drug protein identifiers collected from DrugBank [22] were used to construct a protein vector with a total length of 5,643 items. MedDRA version 16.1 was retrieved from Embl (http://sideeffects.embl.de/download/) and DrugBank version 5.1.1 was retrieved on July 3, 2018 from the DrugBank repository (https://www.drugbank.ca/releases/latest/). A total of 2,591 unique drugs with relevant DrugBank identifiers were collected. From the Merged-PDDI dataset, the total of 297,816 known drug interaction pairs was retrieved. From MedDRA, the number of collected drug adverse effects was 309,849, and there were 6,061 unique adverse effects. The MedDRA data source contains 1,430 unique drugs, but the number of drugs overlapping with the Merged-PDDI dataset source was 995. Therefore, only the adverse effects of 995 drugs were considered during the evaluations.
2. Methods Used in DDI Predictions
In the prediction of potential DDI interactions, the similarities of drugs in known interactions were used. The similarities between drugs were calculated based on factors including drug interaction profile, adverse effect, and drug target. When predicting new interactions, our approach derives DDI predictions based on the degree of similarity between the drug pairs in known DDIs and novel drugs. Our main intuition is that if drugA has a known interaction with drug B, and the similarity between drug B and novel drugC is higher than a certain threshold, then there is a possibility that drugA might have an interaction with drugC. In other words, if two drugs are known to have an interaction and there exists another drug that is similar to one of the drugs in the DDI pair, then it is possible that the third drug might result in a DDI.
For the purpose of calculating drug similarities in the prediction of drug interactions, drug-related information must be extracted and represented in a common vector structure. Fingerprints are a type of vector structure, which were used to represent drug information as drug interaction profiles, proteins, and adverse effects. Before the similarity calculations, the vectors representing the drug IPFs, adverse effect profile fingerprints, and protein profile fingerprints were generated using training data.
When drug IPFs were constructed, each drug was represented by a vector with the length of 2,591. The index “i” has the value of 1 if the current drug interacts with the drug with index “i”; otherwise, it has the value of 0. Similarly, the adverse effect profile fingerprints with 6,061 representing all unique adverse effects were produced for the drugs. For the adverse effect fingerprint of a drug, the value of the index “i” was set to 1 if the drug had an adverse effect with index “n”. Otherwise, the value of the index was set to 0. For instance, the adverse effect vector drug “leucovorin” is populated by 1 at the index 2. The index 2 in the adverse effect vector represents ‘abdominal pain’. This means that the drug leucovorin may have abdominal pain as an adverse effect.
The protein profile fingerprints were also constructed in a similar way. Each drug was represented as a protein vector with the length of 5,643. This vector represents the combination of carrier, target, enzyme, and transport proteins. For each protein, an index is given, and the value of the corresponding entry has the value of 1 if the drug has a relation with this protein. Otherwise, the value is set to 0.
1) Similarity matrix generation
In this section, the similarity matrix calculations to be used for predictions are discussed in detail. To perform predictions, the interaction matrix M1 was constructed based on the fingerprint vectors of drugs. For instance, M1 for the drug IPFs has the size of 2,591 × 2,591. The value of M1(i, j) in the matrix is 1 if the drug with index “i” and the drug with index “j” are known to have a DDI with each other. Otherwise, the value of M1(i, j) is 0.
The similarity matrix M2 is populated based on the similarity between the fingerprint vectors for the drug pairs represented in the interaction matrix M1. The similarity matrix M2 is the same size as M1. The value of M2(i, j) represents the similarity value between a drug with index “i”, and a drug with index “j” and takes a value between 0 and 1 calculated based on similarity measurement. Tanimoto coefficient measurement was used to calculate the similarities in the IPF vectors.
The Tanimoto coefficient, also known as the Jaccard index is a widely used similarity measure that gauges the similarity between finite sample sets [23]. It is calculated as the ratio of the intersection of two sets to the size of their union. It can take values in the range between 0 (minimum similarity) and 1 (maximum similarity). The Tanimoto coefficient is a suitable candidate for measuring similarity between drugs due to its simplicity, reliability, and the interpretability of its range. The similarities are calculated by taking the dot product of interaction profile vectors and applying normalization. In other words, the results are divided by the number of total 1's minus the number of common 1's in these two vectors, and the value is set to M2(i, j). The inner product and Tanimoto coefficient equations are formulated as follows:



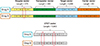
The prediction matrix M3 is calculated by a special cross product of interaction matrix M1 and similarity matrix M2. It is calculated in such a way that the result of product M1(i, j) and M2(j, k) is not summed with the result of product M1(i, j+1) and M2(j+1, k) but saved. In the end, the largest product value is set to M3(i, k). This calculation method is illustrated in Figure 1.
The prediction matrix M3 is used to predict the likelihood of a drug with index “i” interacting with a drug with index “j”. If M3(i, j) has a value greater than 0, then it is considered that the relevant drugs may interact with each other. In the evaluations, M3 was calculated for these three different similarity matrices M2.
2) Calculating Similarities between Protein Vectors
The impact of protein similarities between drugs in DDI was also examined. The degree of protein similarity among drugs with known interactions was explored as a predictor of potential drug interactions. Our intuition is that if drug A and drug B interact with each other, then it is possible that these two drugs might have similar protein fingerprints [6]. That is, when two drugs have high protein similarity, there is a chance that these drugs might have a potential drug interaction.
To evaluate this intuition, the same vector representations used in protein profile fingerprints among drugs with known interactions as well as interaction matrix M1 and similarity matrix M2 were utilized. Not only “approved” protein data but also the protein data under the “all” title in DrugBank were used in evaluations.
Each drug was represented by a carrier target enzyme transporter (CTET) protein vector. A CTET vector is a compound vector that represents a total of 5,643 features with index values of 0 or 1 derived from CTET protein vectors retrieved from DrugBank [22]. For protein identifiers, there were 83 carriers, 375 enzymes, 4,985 targets, and 200 transporters. The value 1 for an index “i” in a CTET protein vector means that the drug has a relation with the protein represented in the corresponding index. An example showing how drugs are represented as protein vectors is shown in Figure 2. The top panel in Figure 2 demonstrates the detailed breakdown of a CTET vector into individual protein vectors, and the bottom panel shows the combined CTET vector representation.
The accuracy of the drug interaction prediction method was evaluated by using known interactions. Then, the drug protein similarities were calculated. The similarity matrix M2 generated for protein profile fingerprints and prediction matrix M3 were used to assess the effectiveness of the approach.
For protein similarity calculations, the Russel Rao similarity measure was utilized. Russell-Rao similarity is a dot-product-based similarity measure in a range between 0 (minimum similarity) and 1 (maximum similarity) [24]. Similar to the Tanimoto coefficient, Russell-Rao represents the normalization of positive matches between vectors. Russell-Rao was chosen for measuring the similarity between drug interactions as it is a straightforward and suitable similarity measure with a 0 to 1 similarity range. That being said, other dot-product-based similarity measures with a 0 to 1 range can also be suitable for vector-based drug-interaction similarities. The Russel Rao similarity is calculated as

In Equation (4), the terms x, y denote binary feature vectors, and d is the vector size. The term xty represents the positive matches between vectors.
3) Evaluation methods
In this study, we performed several evaluations on the collected dataset to assess the performance of the proposed prediction algorithm for different settings. Three different profile fingerprints were used to predict the DDIs in the evaluations, namely, interaction profile fingerprints, adverse effect profile fingerprints, and protein profile fingerprints. For the evaluations, R programming language version 3.5.0 and RStudio version 1.1.419 were used. The receiver operating characteristic (ROC) curves were used to measure the performance of the methods.
III. Results
1. DDI Predictions Based on Profile Fingerprints
1) Interaction profile fingerprint
First, the performance of our DDI prediction approach based on IPFs was evaluated. Out of 297,816 drug interaction pairs, approximately 85.02% of the dataset consisting of 253,201 interaction pairs was selected as training data, and the remaining 44,614 interaction pairs were considered as test data. The values in the prediction matrix were sorted based on the prediction values. Among the top 100 DDI predictions that were not in the training dataset, there were 49 true positive (TP) values.
When the threshold value was set to 0.7, there were 58 TP results among 100 random selections that were not in the training dataset. Similarly, when the threshold value was set to 0.4, the number of TP results was 8. For the test data, the ROC curve was generated as shown in Figure 3. It should be noted that the ROC curves and TP results presented in this paper were in line with those obtained in previous studies [18]. In our evaluations using IPFs, we obtained an ROC value of 0.975 for the 15% test data. It was similar to the ROC of 0.967 found in [8] and 0.98 in [1].
The results showed that the number of TPs was 44,603 among the 44,614 test data. This means nearly all the test DDIs were correctly predicted.
Despite these high TP results, the number of false positive (FP) values was quite large when the threshold value was considered as 0. A total of 2,880,989 FP results were detected among 3,355,345 possible pairs. The total number of possible pairs was calculated by 2591 × 2590 / 2. However, the performance shown in ROC curve indicated that if a certain threshold value higher than 0 was considered, the number of FP results would decrease significantly.
2) Adverse effect profile fingerprint
An adverse effect profile fingerprint vector with 6,061 indexes was used in the similarity calculations because the evaluation dataset consisted of 6,061 unique adverse effects. According to the similarity calculations results, there were 1,675 drug pairs that had similar adverse effect values. This means that there were 1,675 × 2 = 3,350 index values that were greater than 0 in the similarity matrix M2. Based on the prediction matrix M3 results of the test data, we observed that there were 19,644 TP predictions out of 44,614 known test DDI pairs. The number of FPs was 403,765 among all possible 3,355,345 DDI pairs. The ROC curve for the adverse effect profile fingerprint test data is shown in Figure 4.
3) Protein profile fingerprint
Similarly, DDI predictions based on protein profile fingerprints were assessed using the testing dataset. The evaluation results showed that there were 43,430 TP predictions out of 44,614 pairs in the test data. This means that the vast majority of DDI pairs were correctly predicted as potential DDIs. On the other hand, there were 1,965,664 FP predictions among 3,355,345 possible pairs. The ROC curve based on the test data is shown in Figure 5. It should be noted that the ROC curves were similar to those reported in [1].
2. Evaluation of Protein Vector Similarity among Drugs with Known Interactions
The impact of protein similarity in the prediction of potential DDIs were also investigated. We observed that 125,404 pairs out of 297,816 known DDI pairs had protein similarities. In other words, the ratio of TPs was found to be 42%. It is significantly smaller than the TP ratio that was reported in [6], in which it was 72% among drugs with known interactions. The distribution of similarity measurements based on Russel-Rao calculations are shown in Table 1.
On the other hand, the protein similarity distribution ratios were quite similar to those in [6]. As stated in [6], “the higher similarity does not necessarily directly refer to a higher severity of adverse reaction”. More specifically, two drugs with a few common proteins might have a significant DDI, whereas another pair of drugs sharing more common biological elements might not have an interaction. Further, the total number of drug pairs with protein similarities was 224,392 among 3,355,345 all possible pairs. If we consider that 125,404 pairs already had a potential DDI, there were 98,988 pairs that had protein similarities, but their DDI relation is unknown. If we assume that the ratio above 42% is applicable for these unknown pairs, then there might be approximately 41,500 drug pairs with potential DDIs. As seen in Table 2, the protein similarity distribution ratios were very similar to what was shown for known DDI pairs in Table 1.
IV. Discussion
In this study, a DDI prediction approach based on similarities of profile fingerprints was developed. The performance of the approach was experimentally evaluated in detail. The evaluation results indicate that the profile fingerprint approach provides better prediction results compared to the approach of using direct protein similarity among DDI pairs based on TP results. In the profile fingerprint approach, nearly all the test data (15% of known DDI pairs) were successfully predicted with a few exceptions, whereas for the direct protein similarity approach, nearly half of the known interaction pairs were missed. Moreover, the prediction values were positively correlated with the probability of drug interaction for the first approach. On the other hand, the results of the second approach showed that higher protein similarity does not necessarily imply a higher probability of DDI. A drawback of the first approach is that it results in many more FP predictions. When certain thresholds based on ROC curves were considered, the number of FP estimations could be reduced significantly.
For profile fingerprint extraction and prediction, three different types of profiles were evaluated, namely, the drug interaction, adverse effect, and protein fingerprint profiles. Comparisons among those three methods indicated that drug interaction profile method achieved the best results. It was followed by the protein profile method, and then the adverse effect profile method. The similarity-based DDI prediction method using interaction profile vectors successfully predicted nearly all test data with an AUC of 0.975; however, there were still some errors. We think that it is possible to improve the prediction of DDIs by combining prediction methods. In future work, we plan to investigate the feasibility of developing of an improved hybrid prediction approach.
When considering the advantages and disadvantages of the methods, it should be noted that all three methods are based on drug interaction similarities and have the same computational approach; the main difference between the methods is the construction of fingerprint vectors for similarity calculations. The DDI prediction techniques based on drug interaction profiles and protein profiles achieved superior performance, but they produced more FP predictions. However, if certain thresholds based on ROC curves were considered, the number of FP estimations could be reduced significantly.
Furthermore, our evaluations verified that the IPF is a scalable and reliable source of DDI prediction calculation. This means that as the number of known DDI pairs increases and novel drugs emerge, this approach is likely to remain as a reliable DDI prediction model in the future. In comparison to the other two methods, the DDI prediction based on ADEs showed relatively poor performance. Our manual explorations showed that this was mainly due to missing ADEs. Furthermore, the lack of complete overlap between MedDRA and the Merged-PDDI dataset might have had an impact on the performance of the adverse effect profile method because MedDRA does not contain all drugs that are included in the Merged-PDDI dataset.
In conclusion, the results of this study demonstrated that using IPF vector extraction in prediction matrix calculations can achieve good performance in in-silico approaches for DDI prediction.






 XML Download
XML Download