

 PDF
PDF ePub
ePub Citation
Citation Print
Print



MYH9-related disorders (MYH9RD) are rare autosomal-dominant disorders caused by a pathogenic variant of the MYH9 gene. MYH9 consists of 41 exons, the first of which is untranslated. The coding regions of exons 2 to 19 determine the N-terminal globular head (GH) domain, exon 20 determines the neck, exons 21 to 40 determine the coiled-coil (CC) domain, and exon 41 determines the C-terminal non-helical tail of non-muscle myosin IIA. MYH9RD includes four hereditary thrombocytopenias that were previously known as May-Hegglin anomaly (MHA), Sebastian syndrome (SBS), Fechtner syndrome (FTNS), and Epstein syndrome (Table 1). MYH9RD is characterized by macrothrombocytopenia with or without leukocyte inclusion bodies (single or multiple blue or grayish-blue cytoplasmic inclusions located in the peripheral cytoplasm of neutrophils, which are also termed Döhle-like inclusion bodies). MYH9RD is the most common form of inherited thrombocytopenia worldwide. Thrombocytopenia is completely penetrant and characterized by giant platelets, which are larger than red blood cells. The degree of thrombocytopenia is variable and remains stable through life [1]. The presence and severity of bleeding are strongly correlated with platelet count. Approximately 25–30% of patients have spontaneous mucocutaneous hemorrhages, whereas most experience easy bruising and no spontaneous bleeding [1].
MYH9RD should be suspected in patients with thrombocytopenia and giant platelets, with or without family history, since cases from de novo pathogenic variants are common (35%) [2]. In asymptomatic patients, MYH9RD is commonly recognized in adulthood when macrothrombocytopenia is incidentally detected. A significant proportion of MYH9RD patients are initially misdiagnosed with immune thrombocytopenic purpura (ITP) [2]. Awareness of this disease entity and molecular confirmation are important to avoid misdiagnosis and unnecessary treatment, and to manage associated extra-hematological manifestations [3].
In this study, we investigated MYH9 pathogenic variants in five unrelated Korean patients suspected to have MYH9RD.
The patients with macrothrombocytopenia with or without leukocyte inclusions were collected from four institutions. The diagnosis was initially made by hematologists from each institution based on hemato-morphological examinations. We reviewed laboratory data and clinical histories of bleeding tendency, sensorineural deafness, renal impairment, and presenile cataract. The degree of thrombocytopenia was classified according to the automated platelet count at initial diagnosis as mild (100×109/L–149×109/L), moderate (50×109/L–99×109/L), or severe (<50×109/L)[4]. Sensorineural deafness, renal impairment, and presenile cataract were categorized as extra-hematological features. In addition, family members of each proband were examined for the presence or absence of abnormal hemostasis or extra-hematological features.
Blood samples were drawn from the probands and family members for molecular diagnosis of MYH9RD after written informed consent. Genomic DNA was extracted from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit following the standard protocols (Promega, Madison, WI, USA). Direct sequencing experiments were performed to detect variants covering all coding exons and their flanking intronic regions of MYH9 using primer pairs developed by the authors (available upon request). The sequence chromatograms obtained were compared with the reference sequence of MYH9 (NM_0024473.4). Variations identified were described according to the guidelines of the Human Genome Variation Society and classified according to the recent ACMG/AMP guidelines with reference to following public databases: Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genomes (http://browser.1000genomes.org/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), and the Exome Aggregation Consortium (http://exac.broadinstitute.org/).
The clinical characteristics and laboratory findings of the probands and their affected family members (N=10) are summarized in Table 2. The median age of the patients was 26 years (range 6–51). Three patients (30% [3/10]) showed leukocyte inclusions on hemato-morphological examinations. Three patients (30% [3/10]) had bleeding tendency, and two of them had severe thrombocytopenia. One patient (10% [1/10]) had extra-hematological features (high-pitch sensorineural hearing loss and end-stage renal disease). Two patients (20% [2/10]) had been misdiagnosed with ITP, one of whom underwent splenectomy. In all patients, direct sequencing revealed previously reported MYH9 pathogenic variants that were all heterozygous [5].
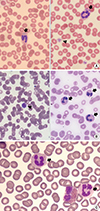
Proband 1 was a six-year-old boy and had severe thrombocytopenia with giant platelets and leukocyte inclusions (Fig. 1A). He had experienced frequent epistaxis once or twice every month since the age of four years. His family members (mother, grandmother, and maternal uncle) had thrombocytopenia with giant platelets but no bleeding tendency. The proband was suspected of MHA, SBS, or FTNS. The pathogenic variant identified was c.5521G>A (p.Glu1841Lys).
Proband 2 was a 39-year-old woman with severe thrombocytopenia with giant platelets and leukocyte inclusions (Fig. 1B). She had no history of bleeding tendency or extra-hematological features. She was suspected of MHA. Her pathogenic variant was c.5521G>A (p.Glu1841Lys). None of her family members had thrombocytopenia.
Proband 3 was a 15-year-old boy who had severe thrombocytopenia with giant platelets and leukocyte inclusions (Fig. 1C). He had no history of bleeding tendency or extra-hematological features. However, his father and uncle had the extra-hematological feature of hearing loss. He was suspected of FTNS. His pathogenic variant was c.99G>T (p.Trp33Cys).
Proband 4 was a 51-year-old woman and had moderate thrombocytopenia with giant platelets. She had no history of bleeding tendency or extra-hematological features. Several of her family members (I-3, I-4, II-3, II-6, and II-7) had macrothrombocytopenia. She and her family members were suspected of MHA. The proband and her affected family members had c.3493C>T (p.Arg1165Cys) (Fig. 2). Her daughter (II-3) was a 30-year-old female and was diagnosed with idiopathic thrombocytopenia and iron deficiency anemia at the age of 23 years. She experienced easy bruising. Other family members (I-3, I-4, II-6, II-7) had no history of bleeding tendency.
Proband 5 was a 22-year-old woman with severe thrombocytopenia with giant platelets. She experienced easy bruising and frequent epistaxis. Audiometry showed high-pitch sensorineural hearing loss. At the age of three years, she had been diagnosed with chronic idiopathic thrombocytopenic purpura and underwent splenectomy. At the age of eight years, proteinuria was detected, which led to end-stage renal disease. She was suspected of FTNS. She had c.287C>T (p.Ser96Leu).
In this study, we describe 10 patients with genetically confirmed MYH9RD (Table 2). Two patients had been misdiagnosed with ITP, one of whom underwent splenectomy. Misdiagnosis of MYH9RD as an acquired form of thrombocytopenia has been reported previously and often leads to delayed diagnosis or administration of ineffective treatment (immunosuppressive drugs and splenectomy) [6]. Collectively with our cases, we reviewed previously reported Korean patients with genetically confirmed MYH9RD [7891011121314]. Data from a total of 43 patients, including 25 probands, demonstrated similar frequency of misdiagnosis: nine patients (20.9%) had been misdiagnosed with ITP, and five of them with severe thrombocytopenia received unnecessary treatment (splenectomy in four). The median age at diagnosis of the series of patients was 24.5 years (range 0–80) with 27.8% (10/36) being older than 39.
For the diagnosis of MYH9RD, identification of family history is helpful, but not necessary, since its absence does not exclude the disease due to frequent de novo pathogenic variants (~35% of probands) [2]. With or without family history, evaluation of peripheral blood smears is a simple and effective diagnostic step in individuals with thrombocytopenia for differential diagnosis of MYH9RD and other inherited thrombocytopenia showing giant platelets from ITP [15]. In addition to macrothrombocytopenia, leukocyte inclusions are a pathognomonic feature specific to MYH9RD. Kunishima et al. [16] and Seri et al. [17] reported leukocyte inclusions in 42–84% of individuals with MYH9RD; these were frequently missed because they were very faint and/or small. Patients with MYH9RD may also have extra-hematological manifestations, such as sensorineural hearing loss, presenile cataract, or proteinuric nephropathy, often evolving to end-stage renal disease [5]. Extra-hematological manifestations of MYH9RD can develop anytime between infancy and adulthood, while macrothrombocytopenia is present from birth. The overall annual rates per 100 affected persons are 1.71 for sensorineural hearing loss, 0.77 for nephropathy, and 0.57 for cataract [1]. Leukocyte inclusions and extra-hematological features were documented in 54.1% (20/37) and 32.6% (14/43) of Korean patients with MYH9RD, respectively. When MYH9RD is suspected because of family history, peripheral blood findings, or extra-hematological manifestations, a molecular genetic test confirms the diagnosis of MYH9RD. Shim et al. [18] also suggested that genetic studies should be done to identify congenital thrombocytopenia in patients who do not recover platelets to greater than 100×109/L in childhood chronic immune thrombocytopenia. Direct sequencing is an effective method for detection of MYH9 gene variants because the disease-causing pathogenic variants are mostly missense variants [5]. Moreover, MYH9RD is known to possess strong genotype-phenotype correlations. The prevalence of spontaneous mucocutaneous bleeding (grade 2 or 3 according to the World Health Organization) and the incidence of extra-hematological features were significantly higher in subjects with MYH9 pathogenic variants in the head domain [119].
All pathogenic variants in our patients were missense variants, and the most common type of pathogenic variants in Korean patients was the missense variant (42/43, 97.7%; Fig. 3A). Ser96Leu (Proband 5) and Glu1841Lys (Proband 1 and Proband 2) were the most common pathogenic variants in Korean patients. Moreover, we noted apparent genotype-phenotype correlations. The incidence of bleeding tendency was higher in subjects with pathogenic variants in the head domain than in those with pathogenic variants in the coiled-coil domain or nonhelical tail (40.0% vs. 15.4% and 0%, respectively, Fig. 3B). The incidence of extra-hematological manifestations was higher in patients with pathogenic variants in the head and coiled-coil domain than in those with pathogenic variants in the nonhelical tail (43.8% and 37.5% vs. 9.1%; Fig. 3C).
In conclusion, data from Korean patients with genetically confirmed MYH9RD demonstrated frequent misdiagnosis as ITP and delayed diagnosis in adulthood. MYH9RD should be suspected in patients with thrombocytopenia, and molecular confirmation can overcome an ambiguous history or findings on peripheral blood smear. In addition, identification of the domain affected by pathogenic variants will help determine genotype-phenotype correlations and genetic counseling.




 XML Download
XML Download