

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
We experienced a case of pigmented paravenous retinochoroidal atrophy (PPRCA), an unusual retinal degeneration characterized by perivenous aggregations of pigment clumps that are distributed along the retinal veins [1]. PPRCA is extremely rare; to our knowledge, only two cases have been reported in Korea [2].
An 8-year-old girl with history of congenital glaucoma and glaucoma surgery reported to our clinic for further evaluation. At the age of five, she had undergone strabismus surgery for exotropia. Her best-corrected visual acuity was 20 / 150 in the right eye and 20 / 33 in the left eye. The anterior segment exam showed a patent peripheral iridectomy site in her right eye. Intraocular pressure, anterior chamber reaction, shape of the optic disc, and pupillary light reflex were all normal. She had right exotropia, but her ocular movement was not limited.
The rod and cone response was detectable but moderately decreased on electroretinography. The fundus showed bilateral bone-spicule pigmentation and retinochoroidal atrophy in the peripheral paravenous distribution without definite macula involvement. No active inflammatory responses of the retina or vitreous, such as vitreous cells, vitreous opacity, retinal infiltration, or hemorrhage, were observed. The Goldmann perimetry test demonstrated a visual field of about 40 to 50 degrees with arcuate scotoma and an enlarged blind spot corresponding to the lesion.
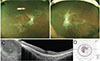
On her last visit, 11 years after the initial presentation, her corrected visual acuity was unchanged: 20 / 200 in the right eye and 20 / 33 in the left eye. There was no significant change in the area of retinal degeneration around the blood vessels, but bone-spicule pigmentation had increased on fundus examination. The area of retinal degeneration was well defined, and diffuse disruption of the outer retina layer was observed in spectral domain optical coherence tomography. The microstructure of the macula remained intact, and the Goldmann perimetry examination was almost stationary (Fig. 1A–1D).
PPRCA has a non-progressive, slow, or subtly progressive course [3]. The etiology of PPRCA is not clearly understood. The majority of PPRCA cases occur sporadically, although there have been specific cases of familial occurrence. Since some patients have bilateral PPRCA with macular coloboma, ophthalmologists have postulated that it is a developmental abnormality and, thus, a disorder of the retinal blood vessels during the embryonic stages of eye development. In the present case, the patient's history of congenital glaucoma suggested that PPRCA may have been caused or accompanied by a developmental problem. On the other hand, a number of inflammatory PPRCA cases has been reported, leading to a different hypothesis citing an inflammatory etiology, including Behcet's disease, measles, rubella, uveitis, and other unknown causes of inflammation [45]. These suggest that PPRCA may be a set of morphologically similar diseases derived from heterogenous causes.
Most PPRCA patients are asymptomatic, although some complain of mild blurred vision. Certain patients, who complain of poor dark adaptation, poor night vision or a blind eye, do not suffer from PPRCA but instead have retinitis pigmentosa (RP) or pseudo PPRCA. PPRCA is commonly bilateral and symmetric. The visual field may be normal, or changes may be variable with topography of pigmentation and atrophy [4].
PPRCA is diagnosed by the typical funduscopic features of bilaterally symmetrical accumulation of pigment and retinochoroidal atrophy along the retinal veins, invariably beginning a distance from the optic nerve head. Peripapillary pigmentary changes may be observed as well as areas of chorioretinal atrophy adjacent to the perivenular pigmentary changes. The unaffected retinal areas appear normal. The pigmentation is typical of bone corpuscle pigmentation, coarse pigment clumps, and fine pigmentary changes. This perivenular appearance is clearly different from RP, which typically shows bony spicule change in the whole retina or a certain sector. As in our case, optical coherence tomography shows degeneration of the outer retina photoreceptor without affecting the inner retina. This characteristic distinguishes PPRCA from vasculitis. Fluorescein angiography, indocyanine green angiography, and electrophysiological tests may be used to confirm the diagnosis [4].
In conclusion, we report a case of PPRCA, a disease that is very rare worldwide. The diagnosis of PPRCA is often neglected due to the paucity of cases and the similarity with RP. Since the disease course and its treatment are different from those of RP or active vasculitis, PPRCA should be distinguished from other similar diseases.
 XML Download
XML Download