

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Alternating hemiplegia of childhood (AHC), rapid-onset dystonia parkinsonism (RDP), and cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) syndrome are distinctive clinical diagnoses that were first described decades ago.123 These syndromes were only recently found to be caused by an ATP1A3 mutation, with the first report of AHC associated with ATP1A3 appearing in 2012.4 Advances in next-generation sequencing techniques have led to rapid expansions of the understanding of the genotypephenotype spectrum associated with ATP1A3 mutations. Several atypical cases with ATP1A3 mutations have recently been identified and have improved the understanding of this condition beyond the above-mentioned distinctive syndromes. However, this has created controversy as to whether these disorders are really distinctive entities or simply part of a broad heterogeneous spectrum, and so the term ATP1A3-related disorders has been proposed.56 ATP1A3-related disorders present with a wide variety of paroxysmal and nonparoxysmal neurological symptoms, and their genotype-phenotype spectrum has not been fully established.
Despite the recent advances in diagnosing ATP1A3-related disorders, only a few treatments have been shown to be useful, and none of them clearly alter the disease course.7 Several treatment modalities for AHC—including flunarizine, topiramate, aripiprazole, and oral adenosine triphosphate—have been described in the literature along with anecdotal reports of their benefits.8 Several recent promising case reports have described reduced disease activity after initiating a ketogenic diet in patients with AHC.91011
This is the first case-series study of patients affected by ATP1A3 mutations in a Korean pediatric population. Our aim was to expand the understanding of the genotype-phenotype spectrum of ATP1A3-related disorders. We also investigated whether a ketogenic diet has therapeutic effects in patients with AHC.
METHODS
Genotype-phenotype spectrum of ATP1A3 mutations
Patients presenting with symptoms compatible with the features of AHC, RDP, or CAPOS were considered for further ATP1A3 testing. The diagnostic criteria and suggestive additional features are summarized in Supplementary Table 1 (in the online-only Data Supplement).12 ATP1A3 sequencing was performed on DNA obtained from probands and family members using the Sanger method after applying the polymerase chain reaction to amplify the entire coding region, the exon-intron boundaries, and the flanking region. Patients with a confirmed ATP1A3 mutation were selected for retrospective chart reviews. The clinical information collected from medical charts included the ages at symptom onset, presentation, and diagnosis, and the family history. Information was collected on paroxysmal symptoms, including symptom semiology, frequency, duration, body area affected, noticeable triggering factors, aggravating/relieving factors, and evolution of symptoms. Information was also collected on nonparoxysmal symptoms, including the presence of persistent motor symptoms such as motor weakness, dystonia, gait disturbance, other movement disorders, and persistent nonmotor symptoms such as intellectual disability and behavioral disorders. Information obtained about the diagnostic workup included EEG, MRI, and metabolic and genetic workups. Detailed information on the treatment history was also collected. ATP1A3 mutation information was collected, and previous reports on identified variants were checked through a literature search. Family testing was performed whenever samples were available from parents. Variants were evaluated and classified according to the guideline proposed by the American College of Medical Genetics (ACMG).13
Ketogenic diet trial
Patients with AHC with a confirmed ATP1A3 mutation phenotype who agreed to try a ketogenic diet were started on the diet. The exclusion criteria included participation in a previous trial of a ketogenic diet for any reason or the presence of other medical conditions that may be adversely affected by such a diet. The parents of those patients who agreed to start ketogenic diet completed a standardized questionnaire to provide a detailed assessment of the current disease severity. The information obtained included the severity and frequency of various paroxysmal symptoms and the degree of the nonparoxysmal manifestations exhibited by the patient.
The patients were admitted for initiation on the ketogenic diet for 5–7 days according to the recommendation of the International Ketogenic Diet Study Group.14 They were first assessed for eligibility to start ketogenic diet by screening for the presence of potential medical factors that may complicate the effects of such a diet. Baseline kidney sonography, echocardiography, and electrocardiography were performed during the admission. The histories and findings of the patients were reviewed. In consultation with a dietitian who specializes in ketogenic diets, the patients began their diet without fasting or fluid restriction. The ketogenic diet was initiated with a fat-to-carbohydrate/protein ratio of 1:1, which was subsequently increased incrementally to a ratio of 3:1. The diet was planned with the goal of a caloric intake of 70 kcal/kg body weight and a protein intake of 1.9 g/kg body weight. The tolerance to the diet was monitored during admission, with the patients evaluated daily for side effects such as dehydration, weight loss, metabolic acidosis, and gastrointestinal complaints. The blood glucose level was measured twice daily to identify potential hypoglycemia, and the urinary ketone level was measured daily. The parents were educated about how to prepare suitable meals for a ketogenic diet. Compliance with the diet and urine ketone levels was checked before discharge.
The primary endpoint of the ketogenic diet was a reduction in the paroxysmal symptom frequency or duration after consuming the diet for 3 months. If the patient experienced a reduction in the paroxysmal symptom frequency or duration of at least 50%, the diet was regarded as effective and was planned to continue consuming it for up to 2 years. The parents were given a standardized symptom check list questionnaire, and the progress of the patients was checked at each visit to the outpatient clinic. The secondary outcome measure was the developmental status and daily functional status of the patients. If the ketogenic diet reduced the paroxysmal symptoms, we planned for the patients to continue the diet and to evaluate their developmental status using the third edition of the Bayley Scales of Infant and Toddler Development. The daily functional status of the patients was screened using the Gross Motor Function Classification System (GMFCS), Manual Ability Classification System (MACS), Communication Function Classification System (CFCS), and Eating and Drinking Ability Classification System (EDACS) at the initial visit and after consuming the ketogenic diet for 3 months.
This study was approved by the International Review Board of Seoul National University Hospital, South Korea (approval no. 1708-115-879), and the parents of all participants provided written informed consent.
RESULTS
Genotype-phenotype spectrum of ATP1A3 mutations
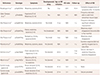
This study recruited 13 patients with confirmed ATP1A3 mutations. Table 1 summarizes their genotype and phenotype spectrum. Ten of these patients showed the AHC phenotype, two patients exhibited RDP, and one patient exhibited CAPOS. The median age of the patients with AHC at disease onset was 6 months (range 0–30 months). Two patients with AHC experienced the neonatal onset of symptoms before 1 months of age. The patients with RDP experienced disease onset at ages of 9 years and 12 years, while this occurred at 7 years of age in the patient with CAPOS syndrome. The median follow-up duration was 106 months (range 27–245 months). The median interval from symptom onset to a genetic diagnosis was 48 months (range 8–360 months). None of the patients had a significant family history.
Paroxysmal symptoms
The most common initial paroxysmal symptom was abnormal ocular deviation in patients with AHC (n=6), followed by motor weakness (n=2) and dystonia (n=2). The median age at the appearance of motor weakness was 12 months (range 6–84 months) in patients with AHC. Both RDP patients presented with acute-onset generalized dystonia with a clear rostrocaudal gradient. Both patients with RDP presented with dysphagia, with one of them also presenting with acutely decreased visual acuity. The patient with CAPOS presented with ataxia, visual impairment, and auditory impairment.
The patients subsequently developed various paroxysmal symptoms. Motor weakness (n=10), abnormal ocular deviation (n=6), seizure (n=2), incontinence (n=2), and ptosis (n=1) were observed only in patients with AHC. Other symptoms such as focal dystonia (n=8), dysphagia (n=7), and dysarthria (n=5) were observed in patients with AHC or RDP, while decreased visual acuity (n=2) was observed in patients with RDP or CAPOS.
The parents of most of the children reported noticeable triggering factors (n=10). A common trigger was a change in body temperature (n=4), such as due to a hot bath, exposure to cold air, or febrile illness such as an upper respiratory infection (n=5). Emotional trigger factors such as anxiety (n=3) or stressors such as fasting or sleep deprivation (n=2) were also described. Sleep (n=8) was a common relieving factor in these patients with AHC. Flunarizine was tried in all 10 AHC patients, which led to partial improvement in either the duration or severity of the paroxysmal symptoms in 6 of them. Topiramate was used as adjunctive treatment in three patients; however, all of the patients who took flunarizine alone or in combination with topiramate continued to experience paroxysmal symptoms (Table 2).
Nonparoxysmal symptoms
The patients showed a wide variety of nonparoxysmal symptoms. All of the patients with AHC showed intellectual disability and developmental delay, which were of varying degrees. Five of the patients with AHC developed persistent neurological deficits. Two patients developed left-arm weakness, two showed persistent dystonia in the upper limbs, and one required support in ambulation due to dystonia in both legs. Treatment with either flunarizine or topiramate had no effect on the nonparoxysmal symptoms of AHC. Both patients with RDP were attending a normal school and achieving average grades until the time of presentation, and exhibited fluctuating but persistent mild focal dystonia. Clonazepam resulted in a modest improvement in dystonia, but without complete resolution. The patient with CAPOS also showed normal development until the time of presentation, but the hearing impairment persisted and required the insertion of a cochlear implant.
Genotype-phenotype spectrum
Two novel mutations were identified in the patients with AHC: p.Ile363Thr and p.Asn743Ser. Mutations that are the most commonly reported in AHC were identified in each of two AHC patients in our cohort: p.Asp801Asn and p.Glu815Lys. One patient with AHC carried the mutation that was previously reported to be associated with the RDP phenotype: p.Arg597Pro. In contrast, the patient with RDP in this cohort carried two mutations that are both reportedly associated with the AHC phenotype: p.Thr769Pro and p.Arg756Cys. The patient with CAPOS had the only mutation that has been associated with CAPOS: p.Glu818Lys.
Most of the patients in this study showed classical features of AHC, RDP, or CAPOS. One exception was patient 4, who showed features of early-onset epileptic encephalopathy at a young age in addition to AHC features. This patient had experienced seizure-like movements since the age of 3 months and had subsequently developed intractable seizures, which were treated with multiple antiepileptic drugs; that patient carried the novel p.Ile363Thr mutation. All mutations were classified as either pathogenic or likely pathogenic according to the ACMG guideline (Table 3).5151617181920
Ketogenic diet trial
Two patients (patients 6 and 8) agreed to proceed with the ketogenic diet trial. Their responses to the ketogenic diet are summarized in Supplementary Table 2 (in the online-only Data Supplement). Patient 6 showed symptoms including abnormal ocular movement starting at 2 months of age, and AHC was diagnosed at the age of 2 years, when he developed recurrent motor weakness. This patient was prescribed flunarizine and topiramate, which did not result in any obvious improvement. The duration of weakness ranged from a few minutes to a few hours, and it occurred once or twice per week. Patient 6 was 41 months old when he began consuming the ketogenic diet. He was not able to stand by himself and could only say a few words (not complete sentences). His daily functional status was GMFCS level 5, MACS level 4, CFCS level 4, and EDACS level 2, and his Bayley score was 85 for cognition (16th percentile, corresponding to a developmental age of 28 months), 68 for language composite (2nd percentile, corresponding to a developmental age 21 months), and 61 for motor composite (0.5th percentile). His gross motor score was affected more than his fine motor score, with developmental ages of 10 months and 27 months, respectively.
Patient 6 was discharged shortly after beginning the ketogenic diet. He was switched from the classic ketogenic diet to a modified Atkins diet (with a fat-to-carbohydrate/protein ratio of 1.5:1) during his initial admission due to feeding difficulties. He remained on the ketogenic diet for 12 months despite the lack of a response at the 3-month follow-up, because he had experienced a symptom-free period of 1 month immediately after initiating the ketogenic diet. However, there were no clear changes in the frequency or duration of paroxysmal symptoms after 12 months, and so the ketogenic diet was stopped. At that time patient 6 was able to stand by holding onto surrounding objects and to speak 30 to 40 words. His daily functional status was GMFCS level 4, MACS level 3, CFCS level 3, and EDACS level 3 at 12 months after the diet initiation. Patient 6 was 5 years old at the last follow-up and could speak two-word sentences.
Patient 8 started to show ocular symptoms at 3 months of age and motor weakness at 10 months of age, and he was diagnosed with AHC after genetic confirmation. He experienced events two or three times monthly, with each event lasting a few days. He was started on a ketogenic diet at the age of 27 months, at which time was able to take only a few steps and could say only a few words. His daily functional status was GMFCS level 4, MACS level 4, CFCS level 5, and EDACS level 2. This patient was admitted and started on the classic ketogenic diet. He was prescribed topiramate and flunarizine, and developed mild metabolic acidosis during the initiation period of the diet. On the third day after diet initiation, the patient suddenly developed a generalized tonic-clonic seizure—he had never experienced any clear electroclinical seizure episode previously. He had repeated seizures on the same day and so phenytoin was initiated, while the ketogenic diet was stopped immediately. He stopped taking phenytoin after 3 months and remained seizure free at the last follow-up. Patient 8 was 45 months old at the last follow-up and could walk alone and speak 10 to 20 words, but not in sentences.
DISCUSSION
The knowledge about the genotype-phenotype spectrum associated with ATP1A3 mutations is increasing as more patients with this mutation are being reported. Several mutation hotspots linked with this specific phenotype have been suggested. AHC is associated with distinctive mutation hotspots in p.Asp801Asn and p.Glu815Lys in about 50% of the reported cases.21 In addition, de novo mutations that cause AHC mainly cluster within or near transmembrane domains. More than 70% of the previously reported mutations are located within transmembrane domains, especially at the M5 and M6 domains, which are nearest to metal-ion-binding sites.22
The current study also identified these mutations as being most frequent in AHC, with 4 of 10 mutations accounted for by p.Asp801Asn and p.Glu815Lys, and 8 of 10 mutations being in close proximity to the M5 and M6 domains.
The p.Glu818Lys mutation has been reported only in patients with CAPOS syndrome, and the patient with CAPOS in the present study also harbored this mutation. An association of ATP1A3 mutation with early-onset epileptic encephalopathy has been reported in two patients with p.Ile363Asn and p.Gly358Val mutations.6 Patient 4 in our study also showed the early-onset epileptic encephalopathy phenotype and carried the p.Ile363Tyr mutation. Given that all of these mutations are located in the P domain, it would be interesting to investigate whether there is a mutation hotspot linked with the epilepsy phenotype. The current case series also found three previously reported mutations that had phenotypes that differed from those in previous reports. Mutation p.Arg597Pro was found in one of our patients with AHC, whereas it was previously reported to be associated with RDP. Mutation p.Thr769Pro was found in our patient with RDP, whereas it was previously associated with AHC. These observations add to the previous reports of phenotype variations despite the presence of the same mutation.23 Mutation p.Arg756Cys was previously associated with an atypical form of RDP or AHC termed relapsing encephalopathy with cerebellar ataxia or fever-induced paroxysmal weakness and encephalopathy.524 However, the patient in the current study carrying the p.Arg756Cys mutation did not experience any acute encephalopathy episode. That patient presented with symptoms more compatible with the classic RDP phenotype, although he also experienced acute transient visual loss, which is atypical for RDP. Therefore, the current genotype-phenotype landscape of ATP1A3-related disorders suggests both a link with specific phenotypes as well as overlapping phenotypes according to the specific mutation or mutation domain. The pathophysiological mechanism for this phenotypic heterogeneity remains to be clearly elucidated. The presence of such wide phenotypic heterogeneity suggests that additional factors such as epigenetic factors or unknown modifier genes play important roles in the clinical expression of ATP1A3-related disorders. However, further research is needed to address this question.
The literature contains some anecdotal reports on patients with AHC improving after initiating a ketogenic diet. The findings of previous studies that evaluated the effects of ketogenic diets are summarized in Table 4.91011252627 Seven of the eight patients reported on previously exhibited favorable outcomes, with no obvious benefit exhibited in the eighth patient. However, the age at the initiation of the ketogenic diet, type of diet regimen, and diet duration varied markedly between the patients. Although these previous studies found mostly favorable outcomes, the evidence is insufficient and needs to be augmented by obtaining more clinical data. Our study attempted to identify any beneficial effects by using a standardized questionnaire for paroxysmal symptoms and by evaluating neurodevelopment outcomes using standardized test tools. The two youngest patients with AHC who exhibited frequent paroxysmal attacks despite receiving treatment with flunarizine and topiramate agreed to try the ketogenic diet. Unfortunately, one of these patients could not continue the diet due to the new onset of recurrent seizures shortly after diet initiation. The other patient did not show meaningful improvement in either the frequency of paroxysmal attacks or neurodevelopment after consuming the diet for 12 months. These negative results question the efficacy of a ketogenic diet in patients with AHC. The current available data suggesting the efficacy of a ketogenic diet were based on anecdotal case reports, and it is possible that cases with negative results have not been reported. The efficacy of a ketogenic diet therefore needs to be reevaluated in a standardized, prospective clinical trial.
In conclusion, this study has further expanded the understanding of the genotype-phenotype spectrum associated with ATP1A3-related disorders. A ketogenic diet, which has previously been reported to exert positive effects in patients with AHC, was not found to be effective in the current study. Evidence to support or disprove the efficacy of a ketogenic diet in patients with AHC remains weak, and so a large-scale systematic clinical trial is needed.



 XML Download
XML Download