Subjects and study design
The study protocol was approved by the institutional review board of Seoul National University Hospital (Seoul, Republic of Korea) and MFDS (
NCT02920047). All the procedures were performed in compliance with the Korean Good Clinical Practice guidelines and tenets of the Declaration of Helsinki. All the subjects provided written informed consent prior to any procedures related to the study.
This study included healthy male subjects between 19 and 50 years of age, weighing ≥ 55 kg and with a body mass index ranging from 18.0 to 27.0 kg/m2. All subjects had no clinically significant abnormalities based on their medical histories, vital signs, physical examination, clinical laboratory tests, and 12-lead electrocardiogram (ECG). Subjects with any hypersensitivity to drugs such as fimasartan and amlodipine were excluded from the study. Additionally, subjects having SBP ≤ 100 mmHg or ≥ 140 mmHg, or DBP ≤ 65 mmHg or ≥ 90 mmHg were excluded from the study at the screening.
This study was designed as a randomized, open-label, two-treatment, three-period, three-sequence, partial replicated crossover study with 14-days washout between periods. The enrolled subjects were randomly assigned to one of the three sequences, and received a single oral dose of an FDC tablet of fimasartan/amlodipine 60/10 mg (Boryung Pharmaceutical Co., Ltd., Seoul, Republic of Korea) as the test drug, or a loose combination of fimasartan 60 mg (Kanarb® tablet 60 mg, Boryung Pharmaceutical Co., Ltd.) and amlodipine 10 mg (Norvasc® tablet 10 mg, Pfizer Inc., Seoul, Republic of Korea) as the reference drug in each period. Each sequence consisted of a single oral administration of the test drug in one period and the reference drug in the other two periods (Sequence A: Reference → Reference → Test; Sequence B: Reference → Test → Reference; Sequence C: Test → Reference → Reference).
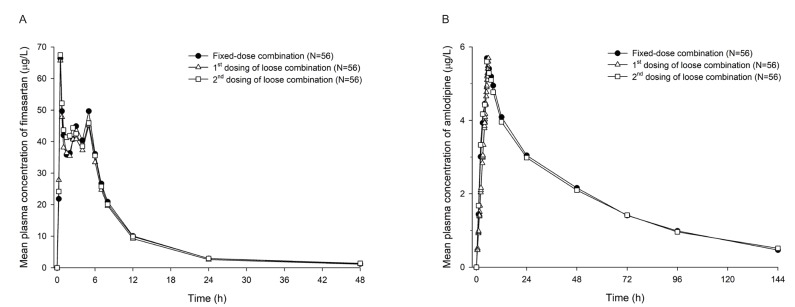
Blood samples for PK analysis of fimasartan were collected at 0 (pre-dose), 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8, 12, 24, and 48 h post-dose. For amlodipine, blood samples were collected at 0 (pre-dose), 1, 2, 3, 4, 5, 6, 7, 8, 12, 24, 48, 72, 96, and 144 h post-dose. Approximately 5 or 8 mL of the blood sample was collected in a heparinized tube for each blood sampling point and subsequently centrifuged at 3,000 rpm for 10 minutes at 4℃. The supernatants were then transferred to three Eppendorf tubes and stored at −70℃ until analysis.
Determination of plasma fimasartan and amlodipine concentrations
Plasma concentrations of fimasartan and amlodipine were analyzed at Kyung Hee Drug Analysis Center of Kyung Hee University (Seoul, Republic of Korea).
Plasma concentrations of fimasartan were determined by a validated high-performance liquid chromatography (HPLC, Agilent 1200 series, Agilent Technologies, USA) coupled with tandem mass spectrometry method (MS/MS, The Applied Biosystems MDS SCIEX API 4000 triple quadrupole mass spectrometer, Applied Biosystems, Canada). In the HPLC system, a Luna C18 column (50 × 2.0 mm, 3.0 µm, Phenomenex, USA) was used for the chromatographic separation of fimasartan and BR-A-563 (Internal standard; IS) under gradient conditions. The MS/MS system was operated in the ionization mode using positive ion electrospray and the multiple reaction monitoring (MRM) mode. The MRM mode was monitored based on an m/z transition of 502.4 → 207.1 for fimasartan and 526.5 → 207.2 for BR-A-563 (IS).
Plasma concentrations of amlodipine were determined by a validated HPLC (Agilent 1100 series, Agilent Technologies, USA) coupled with MS/MS method (The Applied Biosystems MDS SCIEX API 2000 triple quadrupole mass spectrometer, Applied Biosystems, Canada). A Luna C18 column (50 × 2.0 mm, 3.0 µm, Phenomenex, USA) was used for the chromatographic separation of amlodipine and amlodipine-d-4 (IS) under gradient conditions. The MS/MS system was operated in the ionization mode using positive ion electrospray and the MRM mode. The MRM mode was monitored based on an m/z transition of 409.0 → 238.0 for amlodipine and 413.1 → 238.0 for amlodipine-d-4 (IS).
The calibration curves were linear in the range of 1–1000 ng/mL for fimasartan and 0.2–20 ng/mL for amlodipine (r2 ≥ 0.9955 for fimasartan and ≥ 0.9983 for amlodipine). The intra- and inter-batch accuracy ranges were 90.2–106.3% and 99.8–100.6% for fimasartan, and 93.1–105.3% and 99.2–100.0% for amlodipine, respectively. The intra- and inter-batch precision coefficient of variation (CV)% were < 10.0% and < 5.2% for fimasartan, and < 5.5% and < 4.3% for amlodipine, respectively.
PK analysis
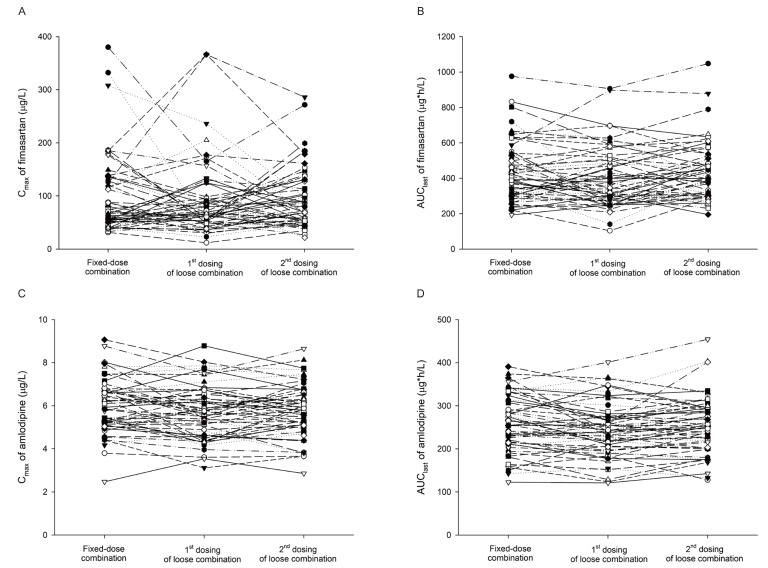
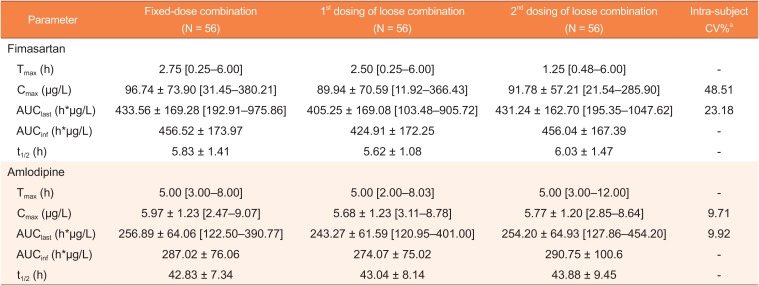
The following PK parameters were calculated by noncompartmental methods using WinNonlin® software, version 7.1 (Pharsight, Mountain View, CA, USA). Cmax and time to reach Cmax (Tmax) were determined from the observed plasma concentration-time profiles. The area under the concentration-time curve from 0 to last measurable time point (AUClast) was calculated using the linear trapezoidal rule for the ascending concentrations and log trapezoidal rule for the descending concentrations. The area under the concentration-time curve from 0 to infinity (AUCinf) was calculated using the following formula: AUCinf = AUClast + Clast/λz, where Clast is the last measurable concentration, and λz is the terminal elimination rate constant. The terminal elimination half-life (t1/2) was calculated as 0.693/λz.
Statistical analysis
A minimum sample size of 45 subjects was estimated to achieve the widened bioequivalence range of the HVD with 80% statistical power at a 5% level of significance, assuming that the highest intra-subject variability of fimasartan was 62%.[
1719] After considering the dropout rate, the total number of 60 subjects were chosen to enroll in this study.
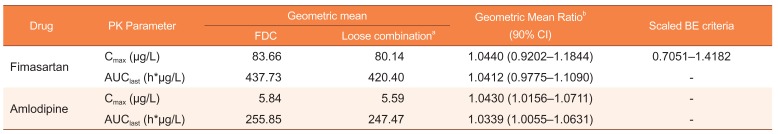
The statistical analyses were performed using SAS® software version 9.4 (SAS Institute, Cary, NC, USA). Analysis of variance (ANOVA) was performed to compare the treatments, considering period, sequence, and the group as fixed effects, and subject nested within the sequence as a random effect. Geometric mean ratios (GMRs) and its 90% confidence intervals (CIs) for C
max and AUC
last variables were estimated. The scaled bioequivalence criteria for the C
max of fimasartan was calculated using exp [±0.760
*(SWR)], where S
WR is the intra-subject standard deviation of the log-transformed values of C
max of the reference drug estimated by this study results.[
1719] The bioequivalence between the two treatments was assessed by using the scaled bioequivalence criteria for the C
max of fimasartan and the conventional bioequivalence criteria for the other PK variables of fimasartan and amlodipine. We used Cochran's Q test to evaluate whether the incidence of adverse events (AEs) is different between the treatments.
Blood pressure monitoring
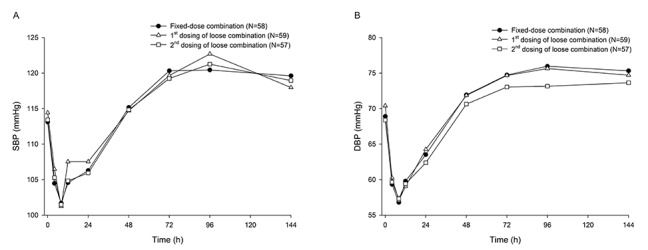
SBP and DBP were measured at 0 (pre-dose), 4, 8, 12, 24, 48, 72, 96, and 144 h post-dose in each period.
Safety and tolerability assessments
Safety and tolerability were evaluated by AE monitoring, clinical laboratory tests, 12-lead ECG, physical examination, and vital signs. All the AEs were coded according to the Medical Dictionary for Regulatory Activities ver.19.1 and summarized by treatment, severity, and relationships with treatments.


 PDF
PDF ePub
ePub Citation
Citation Print
Print



 XML Download
XML Download