

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
After preliminary observations indicated that procainamide's acetylated metabolite, N acetylprocainamide (NAPA), had antiarrhythmic activity in procainamide-treated patients,[1] and pharmacokinetic studies in healthy subjects indicated that NAPA's longer elimination half-life would permit a more convenient dosing interval than recommended for procainamide,[2] patient trials were begun to further develop this metabolite as an antiarrhythmic drug in its own right.[3] As part of the subsequent development program, NAPA pharmacokinetics were investigated in functionally anephric patients in a study that was conducted in continuity before, during, and after hemodialysis.[4] Using a three-compartment mammillary model in which the volume of the central compartment corresponded to plasma volume, it was observed that the intercompartmental clearance to the slowly equilibrating peripheral compartment was reduced during hemodialysis by an average of 77% and only partially returned to normal in the first few hours after hemodialysis. Because the central compartment of the model represented intravascular space, it was concluded that the intercompartmental clearances of the model represented the process of transcapillary exchange.
Identification of model compartments and components of intercompartmental clearance
A similar three-compartment model was used to study the kinetics of simultaneously injected inulin and urea in dogs[5] and in healthy human subjects.[6] The following permeability-flow equation, which has a long history[7] but was first developed by Renkin[8] to describe solute transfer across isolated perfused hind leg capillaries of cats in terms of their blood flow (Q) and permeability coefficient-surface area products (P·S), was then used to interpret the intercompartmental clearance (CLI) results:
Since the transcapillary exchange of inulin is not restricted by molecular size,[9] it was assumed that the ratio of P·S values for inulin and urea would be the same as the ratio of their free-water diffusion coefficients. In this way, simultaneous Renkin clearance equations for urea and inulin, and this ratio were used to calculate values for blood flows and aggregate capillary P·S values for the two peripheral compartments of the model. It was found that the sum of the compartmental blood flows averaged 97% of measured cardiac output (range 83–113%) in dogs,[5] and in humans 5.39 ± 0.49 L/min, similar to the independently measured cardiac output value of 5.47 ± 0.40 L/min.[6] Further confirmation of the validity of this approach was provided by a pharmacokinetic study in dogs in which inulin, urea, and theophylline were injected simultaneously.[10] Theophylline kinetics were modeled with a similar three-compartment model, and its intercompartmental clearances were similar to the blood flows estimated for the corresponding inulin and urea compartments. The anatomic identity of the peripheral model compartments was then explored by studying the pharmacokinetics of these three compounds in dogs after splenectomy and enterectomy.[11] Because this surgical procedure removed approximately half of the volume of the rapidly equilibrating but less than 20% of their slowly equilibrating inulin and urea peripheral compartments, it was concluded that the fast equilibrating compartment was composed primarily of splanchnic tissues, and the slowly equilibrating compartment was made up primarily of skeletal muscle and other somatic tissues. Based on previous morphologic studies, it was inferred that the difference in transcapillary exchange rates of the peripheral compartments reflected the fact these three compounds reached the interstitial fluid of splanchnic organs primarily by diffusing through the relatively large fenestrations of splanchnic capillaries, whereas they reached somatic tissues through the much narrower endothelial cell junctions of continuous capillaries.
Studies in a canine hemodialysis model
This modeling approach subsequently was used to analyze inulin and urea distribution kinetics before, during, and after hemodialysis of dogs with intact kidneys.[12] During the two-hour hemodialysis session, mean arterial pressure fell by less than 5%, but total peripheral resistance increased by 47%, and cardiac output fell by 35%. These hemodynamic changes were accompanied by marked reductions in the slow CLI of inulin and urea to 19% and 63% of their respective predialysis values. Using the simultaneous Renkin equation approach, it was estimated that this corresponded to a 90% reduction in blood flow to this compartment during hemodialysis. These changes were only somewhat attenuated during the two hours after hemodialysis. In contrast to these marked changes, the P·S values for this compartment were not significantly reduced, signifying that there was no significant derecruitment of capillaries supplying skeletal muscles and other somatic compartment components. There also was no significant change in the blood flows or P·S values calculated for the more rapidly equilibrating splanchnic compartment.
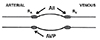
The additional observation was made that these changes were accompanied by activation of the renin-angiotensin system. The stability of somatic compartment P·S values during hemodialysis was in marked contrast to what was observed in the previous study in which splanchnic tissues were removed to assist in compartment identification.[11] In this study, there was a marked reduction in slow compartment P·S values that was correlated with a surgery-induced elevation in plasma levels of arginine vasopressin (AVP) such that AVP levels of 45 pg/ml resulted in an almost 50% decrease in P·S values.[13] The observed reduction in P·S values results from the fact that AVP acts solely to increase precapillary arteriolar resistance and so lowers intracapillary pressure to the extent that functional capillary density decreases and aggregate capillary surface area is reduced (Fig. 1).[14] On the other hand, angiotensin II (AII) acts in a balanced fashion to constrict both precapillary and postcapillary resistance vessels in such a way that sufficient intracapillary hydrostatic pressure is retained to maintain patency.[15]
Subsequent clinical studies
Although kinetic studies in both hemodialyzed patients and dogs indicated that this procedure was accompanied by substantial hemodynamic changes that were likely related to the occurrence of skeletal muscle cramps, they failed to provide evidence as to why only some hemodialyzed patients experienced these cramps. The study of hemodynamic and hormonal responses to hemodialysis is complicated by the concurrence of both osmotic and volume stimuli during the procedure and by the possible dialytic removal of vasoactive substances. Because of the role played by plasma volume contraction in initiating intradialytic cramps, head-up tilt was selected as a relatively well-controlled volume stress that could be used for comparing the responses of 8 patients who frequently experienced intradialytic cramps with those of 8 patients who cramped infrequently.[16] Baseline blood pressure, plasma renin activity and plasma norepinephrine and arginine vasopressin concentrations were similar in both groups. After equilibration in the recumbent position, the patients were brought to a 60° tilt for up to 1 hr. Systolic blood pressure rapidly fell by an average of 17% in patients who cramped infrequently but only by an average of 10% in frequently cramping patients. The ratio of tilt/recumbent norepinephrine levels was correlated with the extent to which plasma volume was decreased and exceeded 1.5 in 7 patients with frequent cramps but was less than 1.2 in 6 patients who cramped infrequently. Tilting also increased plasma AVP levels in the patients with frequent cramps but did not increase plasma renin activity significantly in this patients' group.
Because norepinephrine is like AVP in that it acts solely to increase precapillary arteriolar resistance,[15] we inferred that activation of the sympathetic nervous system during hemodialysis was responsible for reducing intracapillary pressure to the extent that capillary derecruitment led to skeletal muscle ischemia and cramping (Fig. 1). Of further interest is that one infrequently cramping patient whose norepinephrine response more than doubled during tilting also had a normal plasma renin response to tilt. In this respect, this patient was similar to the dogs with intact kidneys and renin-angiotensin system who showed no evidence of capillary derecruitment, as evidenced by the stability of their somatic compartment P·S values. On the basis of these observations, we concluded that skeletal muscle cramps were occurring in some hemodialysis patients because their renal production of renin had decreased in parallel with the deterioration in their renal excretory function. For that reason, these patients did not generate a sufficient quantity of AII to modulate the sympathetic nervous system activation that occurred in response to the volume stress of hemodialysis. On the other hand, most patients who cramped infrequently had a deficient sympathetic nervous system response to volume stress since norepinephrine levels have been found to usually rise by 50% after 10 minutes when healthy subjects are tilted.[17]
These findings led to a hypothesis regarding the pathophysiology of intradialytic skeletal muscle cramps that is summarized in the flow chart shown in Figure 2. Based on this hypothesis, one way to prevent intradialytic cramps would be to reduce the sympathetic activation that occurs in response to the volume stress resulting from hemodialysis. Quinine was used for many years to mitigate intradialytic cramps following demonstration of its efficacy in a randomized, double-blind, crossover trial.[18] The efficacy of quinine in this regard probably reflects mainly the sympatholytic effect that results from its α-adrenergic blocking activity.[19] Subsequently, it was demonstrated that the α-adrenergic blocking drug prazosin was also effective in reducing intradialytic cramp frequency.[20] However, the hypotensive effects of this drug limit the magnitude of the doses that can be administered and make administration of even small doses hazardous without close patient monitoring. Nonetheless, these studies do buttress the hypothesis diagrammed in Figure 2. Further support is provided by a small double-blind pilot study in which captopril was administered to five patients in order to rule out the possibility that excessive activation of the renin-angiotensin system was responsible for their intradialytic cramps. [21] Predialysis administration of captopril actually increased intradialytic cramp frequency in one patient but had no effect on cramping in the other four patients.
Because renin-angiotensin system function is attenuated in patients with end-stage kidney disease who are being treated with hemodialysis, the most physiologically consistent approach to preventing their intradialytic cramps would be to administer AII to them while they are being dialyzed. This is conceptually similar to the administration of insulin to patients with type I diabetes and could be accomplished by using the venous port of dialysis machines for intravascular administration. Intravenous infusions of AII have been used successfully to treat patients with a distributive shock that did not respond to norepinephrine, and AII is marketed for this indication.[22] Unfortunately, the small number of patients with intradialytic skeletal muscle cramps and the logistical difficulties in obtaining reimbursement in hemodialysis patients have discouraged the commercial development of adjuvant therapies that might benefit them.

 XML Download
XML Download