

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Primary immunodeficiency diseases (PIDs) are a heterogeneous group of inherited disorders of the immune system. More than 300 genetic defects have been detected as causes of PIDs.1 The estimated prevalence of PIDs has been reported to be 2.3 per 100,000 persons in Japan,2 while that in Korea is 1.125 per 100,000 children based on questionnaire surveys in 2012.3 However, recent reports suggest that PIDs are more common than previously believed, and the prevalence of any diagnosis in the US increased from 38.9 to 50.5 and 29.1 to 41.1 per 100,000 among publicly and privately insured persons, respectively, between 2001 and 2007.4 In addition, during the 10 years from 2004, which the European Society of Immune Deficiencies (ESID) online registry system has been operational, the number of patients registered has grown substantially; after 4 years, more than 7,000 patients were registered and, as of 25 June 2014, 19,355 patients in Europe have been registered as having a PID.5
PIDs have been diagnosed using the ESID registry (2014) criteria6 based on clinical manifestations and hematological/biochemical laboratory tests. The clinical phenotypes of patients are heterogeneous; therefore, the accurate identification of an underlying PID may not always be possible. The appropriate diagnosis of PIDs is crucial for initiating proper management and reducing infection-associated morbidity and mortality, and therefore, detection of specific protein abnormalities or significant genetic variants is crucial. Particularly, molecular tests are essential for identifying the structural abnormalities of genes, thus enabling accurate diagnosis.78 However, genetic tests are time-consuming, laborious and costly. Moreover, targeted single gene tests may miss causative mutations. Exome sequencing has recently been received attention as a diagnostic tool for many rare genetic diseases, including PIDs. However, it has been reported that exome sequencing detected causative genetic defects in about 40% of patients with PIDs with variable size of panels.910 Exome sequencing is still limited as a diagnostic test because of the high cost, labor intensity and turn-around time. Flow cytometry (FCM) is a diagnostic tool used for both immunophenotyping and functional assays of immune cells in real time. FCM has been used to assess functional characteristics linked to PIDs.11 Thus, it can be an initial screening test for rapid diagnosis in a spectrum of patients suspected of having PIDs. For example, the absence of T cells, natural killer (NK) cells or both in severe combined immunodeficiency (SCID), absence of B cells in agammaglobulinemia, or markedly reduced dihydrorhodamine (DHR) in X-linked chronic granulomatous diseases (XL-CGD) provide very significant evidences of PIDs and should draw the immediate attention of clinicians to start therapeutic intervention while awaiting results of confirmatory genetic tests.
In this study, we retrospectively reviewed the use of FCM tests for the diagnosis of PIDs at a single tertiary care center in Korea. The aims of this study were as follows: 1) to highlight the experiences of a diagnostic approach for patients with PIDs using FCM, and 2) to suggest FCM tests, based on positive findings as a first line diagnostic approach and treatment monitoring tool in certain PID groups.
MATERIALS AND METHODS
Patients
Between January 2001 and June 2018, 115 patients who were highly suspected of having PIDs were subjected to FCM or genetic tests or both at the Samsung Medical Center, Seoul, Korea. After exclusion of 27 patients with secondary hemophagocytic lymphocytic histiocytosis, 8 carriers, and 20 patients who did not meet the ESID criteria (2014), 60 patients were included in this study (Fig. 1). Clinical manifestations and laboratory findings were collected from electric medical records. The study was approved by the Institutional Review Board (IRB) of the Samsung Medical Center (IRB No. 2019-07-175-003) and performed in accordance with the principles of the Declaration of Helsinki.
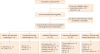 | Fig. 1Enrollment and classification of patients.PID, primary immunodeficiency disease; HLH, hemophagocytic lymphohistiocytosis; ESID, European Society of Immune Deficiencies; IUIS, International Union of Immunological Societies; WAS, Wiskott-Aldrich syndrome; AD-HIES, autosomal dominant-hyper-IgE syndrome; HIES, hyper-IgE syndrome; BTK, Bruton's tyrosine kinase; CVID, common variable immune deficiency; IgA, immunoglobulin A; CTLA4, cytotoxic T-lymphocyte associated protein 4; ALPS, autoimmune lymphoproliferative syndrome; AR-CGD, autosomal recessive chronic granulomatous diseases; FHL, familial hemophagocytic lymphohistiocytosis; LAD, leukocyte adhesion deficiency; SCID, severe combined immunodeficiency; IPEX-like SD, immunodysregulation polyendocrinopathy enteropathy X-linked syndrome; XL-CGD, X-linked-chronic granulomatous diseases; STAT1, signal transducer and activator of transcription 1; GOF, gain-of-function.
|
Multicolor flow cytometry
Whole blood samples were collected from patients. The cells were incubated with the surface marker antibodies for 30 minutes at 4°C and washed twice with phosphate-buffered saline (PBS). Red blood cells were lysed using fluorescent activated cell-sorting (FACS) Lysing Solution (Becton Dickinson, San Jose, CA, USA). For intracellular staining, the cells were incubated with a fixation and permeabilization mixture (eBioscience, San Diego, CA, USA) on ice for 1 hour. Then, the cells were washed with PBS and incubated with the antibodies at 24°C for 15 minutes. After the final wash, the cells were resuspended in PBS and FACS acquisition was performed. The neutrophils were activated with phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich, St. Louis, MO, USA) for 15 minutes for the DHR assay, while the lymphocytes were similarly treated for the cluster of differentiation (CD) 11b/CD18 and CD40/CD40L tests. For phospho-flow analysis of the intracellular phosphorylated signal transducer and activator of transcription 1 (pSTAT1), peripheral blood mononuclear cells were acquired by density gradient centrifugation using Ficoll (General Electric Healthcare Bio-Sciences, Uppsala, Sweden) and stimulated with interferon (IFN)-γ. For the DHR and pSTAT1 assays, the stimulation index was calculated for gated neutrophils and lymphocytes by dividing the median fluorescent intensity (MFI) of stimulated cells with the MFI of unstimulated cells. The flow cytometer and analysis software used were FACSCanto II (Becton Dickinson) and FACSDiva ver. 8.0 (Becton Dickinson), respectively. Immunophenotyping was performed using the panels of antibodies depending on the suspected PIDs. The unstained cells and cells stained with the relevant isotype antibodies were used as controls. Detailed information about the antibodies and clones is provided in Table 1.
Table 1
Flow cytometry panels for primary immunodeficiency disease diagnosis

BTK, Bruton's tyrosine kinase; ALPS, autoimmune lymphoproliferative syndrome; DHR, dihydrorhodamine; HLH, hemophagocytic lymphohistiocytosis; CD, cluster of differentiation; TCR, T-cell receptor; STAT1, signal transducer and activator of transcription 1; pSTAT1, phosphorylated signal transducer and activator of transcription 1; NADPH, nicotinamide adenine dinucleotide phosphate; CCR7, C-C chemokine receptor type 7; IgD, immunoglobulin D.
![]()
Sanger and exome sequencing
Genomic DNA was extracted from peripheral blood leukocytes using the Wizard Genomic DNA purification kit (Promega, Madison, WI, USA) according to the manufacturer's instructions and each gene mutation analysis was performed on the archival samples. The primers were designed in-house. Direct sequencing was performed using the ABI Prism 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems). The results were analyzed using the Sequencing Analysis software ver. 5.2 (Applied Biosystems). Exome sequencing was performed using the Illumina TruSight panel with the MiSeq HiSeq 2500 platform (Illumina, San Diego, CA, USA). The data were analyzed using the pipeline software programs, which were optimized in the Green Cross Genome Laboratory.
Data analysis
We categorized the utility of FCM tests for clinical diagnosis of PIDs. Depending on the comparability between FCM and genetic diagnosis, it was categorized as providing strong evidence, probable evidence or helpful clues (Fig. 2). Furthermore, where the FCM test was useful for monitoring the treatment response of PIDs is also reported. When FCM could provide evidence to diagnose specific PIDs, we categorized as strong. Probable evidence provided altered findings by FCM for highly suspecting PIDs but rather for broader categories of PIDs, so the molecular test was required. The helpful clue category included PIDs where the FCM findings suggested immunological abnormalities but were not specific for diagnosis and providing clues of treatment responses.
 | Fig. 2Categorization of PIDs based on the usefulness of Flow cytometry for diagnosis.PID, primary immunodeficiency disease; FCM, flow cytometry; AD-HIES, autosomal dominant-hyper-IgE syndrome; ALPS, autoimmune lymphoproliferative syndrome; AR-CGD, autosomal recessive-chronic granulomatous diseases; CTLA4, cytotoxic T-lymphocyte associated protein 4; CVID, common variable immune deficiency; FHL, familial hemophagocytic lymphohistiocytosis; IPEX-like SD, Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome; LAD, leukocyte adhesion deficiency; SCID, Severe combined immunodeficiency; WAS, Wiskott-Aldrich syndrome; XIAP, X-linked inhibitor of apoptosis protein; XLA, X-linked agammaglobulinemia; XL-CGD, X-linked-chronic granulomatous diseases.
*FCM was not performed for these diseases in this study; †We suggest that WAS could be divided into 2 categories based on the variants that influence protein expression.
|
RESULTS
Strong evidence: SCID, X-linked agammaglobulinemia (XLA), XL-CGD, leukocyte adhesion deficiency (LAD), autoimmune lymphoproliferative syndrome (ALPS)-FASLG and familial hemophagocytic lymphohistiocytosis (FHL)
Six patients had experienced frequent pneumonia, lung abscess, or orbital cellulitis at 3–7 months of age. Their lymphocyte subset results showed that the T cells were markedly decreased to 0%–4% (Supplementary Fig. S1), which suggested that these patients had issues with T cell generation and differentiation (Table 2). All the patients were presumptively diagnosed with SCID. Among them, 4 were confirmed to have γ chain (γc) deficiency due to the IL2RG gene mutation, whereas the others did not proceed to take genetic tests but they could have started the treatment based on the FCM results. Of the 11 patients finally diagnosed with Bruton's XLA due to the BTK mutation (Table 3), all 5 who were subjected to lymphocyte subset analysis had almost no circulating CD19+ B cells. Two of the five patients tested for intracellular Bruton's tyrosine kinase (BTK) protein expression on monocytes showed markedly reduced expression (Supplementary Fig. S2), whereas the other 3 having TTTG deletion in intron 15 showed moderately reduced expression. Four XLA patients referred from other hospitals for therapeutic options, including intravenous immunoglobulin (IVIG) and hematopoietic stem cell transplantation (HSCT), were not subjected to FCM and were diagnosed only using genetic tests.
Table 2
FCM and genetic test findings of severe combined immunodeficiency patients (n = 6)

FCM, flow cytometry; M, male; NT, not tested; CD, cluster of differentiation; NK, natural killer; XLR, X-linked recessive. *Age when intravenous immunoglobulin started.
![]()
Table 3
Flow cytometry and genetic test findings of X-linked agammaglobulinemia patients (n = 11)

FCM, flow cytometry; CD, cluster of differentiation; BTK, Bruton tyrosine kinase; Control, healthy control; M, male; NT, not tested; XLR, X-linked recessive.
*Age intravenous immunoglobulin was started. †Outside means that the genetic tests had been performed in referred hospital and the detailed information of genetic tests were not provided.
![]()
Four of the 11 patients with XL-CGD had deep seated infections, including liver abscesses (Serratia marcescens, n = 1), splenic abscesses (Candida species, n = 1), osteomyelitis (n = 1) and cellulitis (Burkholderia cepacia, n = 1), which implied susceptibility to bacterial infections (Table 4). Four patients presenting with post-Bacille Calmette-Guérin lymphadenitis (n = 2), tuberculosis lymphadenitis (n = 1), and pneumonia with pleural effusion and hepatosplenomegaly (n = 1).12 Three patients had no clinical records at our center. The nicotinamide adenine dinucleotide phosphate (NADPH) oxidase function measured by the DHR tests in these 11 male patients showed profound abnormalities, such as having almost no respiratory burst activities (Supplementary Fig. S3). These findings strongly suggested a diagnosis of XL-CGD. Subsequently, a CYBB gene mutation was detected in all 6 patients subjected to genetic tests.
Table 4
Flow cytometry and genetic test findings of X-linked chronic granulomatous diseases patients (n = 11) and leukocyte adhesion deficiency patients (n = 3)

FCM, flow cytometry; AR, autosomal recessive; Control, healthy control; DHR, dihydrorhodamine; CD, cluster of differentiation; F, female; M, male; NT, not tested; XLR, X-linked recessive; MFI, median fluorescence intensity.
*MFI ratio: median MFI of stimulated DHR test/median MFI of unstimulated DHR test; †The result of CD18/CD11 is presented as a percentage (%); ‡Outside means that the genetic tests had been performed in referred hospital and the detailed information of genetic tests were not provided.
![]()
The 3 patients who manifested omphalitis, otitis media or mastoiditis were subjected to CD18/CD11b LAD panel tests (Table 4). The results of the FCM showed the absence of cell surface CD18 expression without and with stimulation, which strongly suggested a presumptive diagnosis of LAD1 (Supplementary Fig. S4). Two patients who had been subjected to genetic tests had mutations in the ITGB2 gene.
We had a 5-year-old patient who had experienced recurrent pneumonia, intestinal lymphangiectasia, and bacterial peritonitis induced by Escherichia coli. The lymphocyte subset analysis showed increased CD4+ T cells, but the ratio of CD4+/CD8+ T cells was within the reference range. However, the CD4 and CD8 double negative T-cell receptor (TCR)-αβ+ T cells (DNTs) were significantly increased to 6.9%. The presumptive diagnosis of ALPS was confirmed by a genetic test showing the heterozygous mutation of the FASLG gene [FASLG:c.436G>T, p.(Val146Leu)].
A 29-day-old female infant, the second baby born in full term (38 + 4 weeks, 3,500 g), was admitted because of mild fever and rapid breath. Her sister had died from sepsis, acute hepatitis, gastrointestinal bleeding, and pulmonary hemorrhage at 28 days of age. The result of her laboratory tests met the hemophagocytic lympho histiocytosis criteria 2004. The FCM test for intracytoplasmic perforin expression showed the complete absence of perforin expression in her NK cells and CD8+ T cells. However, her parent was healthy and had a comparable expression of perforin (data not shown). Subsequently, deleterious biallelic mutations of the PRF1 gene inherited from each parent were detected [PRF1:c.65delC, p. (Pro22Argfs*2), and c.1090_1091delCT, p.(Leu364Glufs*93)], which confirmed FHL2.
Probable evidence: autosomal recessive-chronic granulomatous disease (AR-CGD), autosomal dominant-hyper-IgE syndrome (AD-HIES) and signal transducer and activator of transcription 1 (STAT1) gain of function (GOF) mutation
Compared to XL-CGD, the respiratory burst activities of 2 AR-CGD patients identified having NCF1 and CYBA mutations were moderately reduced compared to healthy controls (Table 5). Both patients had histories of recurrent infections, including a lung abscess by Aspergillus fumigates and multiple fungal abscesses on the retroperitoneum and liver by Candida krusei.
Table 5
Flow cytometry and genetic test findings of AR-chronic granulomatous disease patients (n = 2), autosomal dominant hyper-IgE syndrome (n = 1) and STAT1 mutation (n = 1)

FCM, flow cytometry; AD, autosomal dominant; AR, autosomal recessive; Control, healthy control; DHR, dihydrorhodamine; IL, interleukin; pSTAT1, phosphorylated signal transducer and activator of transcription 1; F, female; M, male; MFI, median fluorescence intensity.
*MFI ratio: median MFI of stimulated DHR test/median MFI of unstimulated DHR test; †The result of pSTAT1 and IL17 were presented in percent (%); ‡Outside means that the genetic tests had been performed in referred hospital and the detailed information of genetic tests were not provided.
![]()
A patient was diagnosed with AD-HIES induced by the loss of function (LOF) mutation of STAT3. This patient had presented with bronchiectasis, nontuberculous mycobacteria (NTM) pulmonary infection, and recurrent skin infections. The intracellular staining of interleukin (IL)-17 in PMA/ionomycin-activated CD4+ T-cells showed an almost absence of IL-17-secreting CD4+ T-cells (0.01% in patients vs. 2.36% in controls), which suggested abnormalities of the STAT3 signaling pathway.
The 24-year-old female patient who had NTM and a recurrent superficial fungal infection had been referred13 for PID evaluation. In exome sequencing, a GOF mutation of STAT1 (c.800C > T) was detected. There was no abnormal finding in initial lymphocyte subset analysis, but the FCM analysis of intracellular pSTAT1 without and with stimulation by IFN gamma showed significantly increased expression of pSTAT1 without inhibition by the Janus kinase (JAK) inhibitor, ruxolitinib (Novartis Pharmaceutical Corporation, East Hanover, NJ, USA).
Helpful clues and utility for treatment monitoring: PIK3CD and CTLA4 mutations and undetermined common variable immune deficiency (CVID)
The patients belonging to this category had abnormal lymphocyte subset results, including a slight to marked decrease in CD19+ B cells, decreased NK cells, or decreased regulatory T cells (Tregs) (Table 6). Female siblings, 24 and 20 years old, had been identified, using exome sequencing, as having an autosomal dominant CTLA4 mutation, after the initial diagnosis of CVID based on clinical findings and decreased B cells 10 years prior. The older sister had exhibited clinical manifestations such as thrombocytopenia, recurrent parotitis, hepatosplenomegaly and ulcerative colitis (chronic diarrhea and hematochezia) from birth and the younger sister was found to have hypogammaglobulinemia (immunoglobulin [Ig] G, 347 mg/dL; reference 700-1,600 mg/dL) at 13 years of age. IVIG medication was commenced following the diagnosis of CVID. Even after IVIG treatment, both siblings experienced multiple episodes of illness, including anemia, thrombocytopenia, viral infection, and malignancies such as gastric cancer and myelodysplastic syndrome. Both patients had markedly decreased B-cells, and the older sister had a decreased CD4+FOXP3+ Tregs (1.8% CD4+ T cells in the patient as compared to 4.9% in the control) (Supplementary Fig. S5). Their mother has the same mutation but did not have clinical symptoms and had normal B- and T-cell proportions.
Table 6
Flow cytometry and genetic test findings of CTLA4 mutation patients (n = 3), PIK3CD mutation patients (n = 2) and undetermined common variable immune deficiency patients (n = 3)

FCM, flow cytometry; CD, cluster of differentiation; NK, natural killer; AD, autosomal dominant; F, female; M, male; NT, not test.
![]()
Two patients presenting lymphadenopathy and recurrent infections, such as acute otitis media and pneumonia, were diagnosed with a PIK3CD GOF mutation by exome sequencing. One patient was found to have a cervical lymph node abscess and lymphocyte infiltration in the colon. In these patients, the usual lymphocyte subset analysis did not show abnormal findings, but a more detailed analysis of naïve and memory T-cell subsets showed altered differentiation status of CD4+ and CD8+ T cells. Their initial analyses showed increased naïve B cells (CD19+IgD+ CD27− cells) and memory cytotoxic T cells (CD3+ CD8+ CD45RA cells) compared to age-matched values (Supplementary Fig. S6).14 After treatment with sirolimus, a mammalian target of rapamycin (mTOR) inhibitor, the altered status of T and B lymphocyte differentiation was improved in these patients with activated PIK3CD mutations.
Three patients were diagnosed with CVID based on clinical manifestations of recurrent infections, including pneumococcal pneumonia, pseudomonas sepsis, and lymphadenitis. Their lymphocyte subset analysis showed variably decreased B cell counts (0%–9%). Although genetic diagnosis by extensive molecular tests such as exome sequencing had not been performed yet, all the patients were under the IVIG therapy based on the ESID diagnostic criteria and the lymphocyte subsets were monitored.
DISCUSSION
We described the usefulness of FCM tests in diagnosing or deciding to start treatment in PIDs or both based on the experience at a single tertiary care hospital. In recent years, FCM has emerged as an extremely useful laboratory tool for evaluating patients with potential PIDs.15 Based on the 2017 International Union of Immunological Societies, genetic tests should be performed to confirm the diagnosis. However, in some cases such as XL-CGD and BTK that have a specific functional assay and a specific protein that can be detected, the FCM methodology could be an efficient laboratory tool to establish a specific diagnosis as well as monitor treatment responses. Moreover, performing lymphocyte subset analysis, which includes analyzing T, B, NK cells, and helper and cytotoxic T cells, provides the initial clue for insufficient immune system.
SCID is a group of diseases caused by genetic defects producing blockade or impairment of differentiation T-lymphocytes and, to a varying degree, other lymphocytes or cells of the myeloid lineage.16 In addition to the clinical phenotypes of SCID, distribution and absolute count of T, B, and NK cell subsets can provide clues regarding the extent to which the immune system is affected.17 The most common form affecting approximately 30%–60% of the patients is X-linked SCID caused by mutations in the gene encoding the common γc of the IL-2R1819 which is mostly presented as T-B+ NK− SCID. Similarly, in our study the IL2RG mutation was found in 3 patients, who were subjected to genetic tests and were almost devoid of CD3+ T-cells (0%–3.2%) and NK cells (0%–4.5%). One patient who had T-B+ NK+ SCID (patient 6 in Table 2) may have a genetic defect such as in IL-7Rα, CD3δ, CD3ε, or CD3ζ although IL2RG and JAK3 mutations have also been shown to cause this phenotype.20 Now, allogeneic HSCT is the only curative option for SCID.21
XLA is characterized by decreased serum immunoglobulins of all isotypes and an almost complete absence of B-cells due to an arrest in B lymphocyte development. Intracellular BTK protein expression in XLA was shown to be reduced or absent.2223 All 5 XLA patients who were subjected to the lymphocyte subset analysis showed an almost complete absence of CD19+ B cells (0.0%–0.1%); however, their BTK protein expression was variable. Two patients had significantly low (15.7% and 2.8%) levels but the other 3 patients who had 4 nucleotide deletions in intron 15 of the BTK gene (BTK:c.1567-12_1567-9delTTTG) in common showed slight to moderate decrease (45.6%–88.6%) in BTK protein expression, and, in them, the expression of the BTK protein seemed to be affected to a lesser extent.
A DHR test measuring the neutrophil respiratory burst activities by NADPH oxidase function is a relatively quick and robust assay to diagnose and determine the potential genotype of CGD.24 In our patients, XL-CGD caused by a CYBB mutation had almost no neutrophil oxidative burst; however, AR-CGD by NCF1 had a partial defect, which was similar with the result of a previous report.2225
Likewise, PIDs caused by the LOF genetic mutation of the ITGB2 gene in LAD-1, the PRF gene in FLH2, and the STAT3 gene in AD-HIES showed absence or a marked decrease of the corresponding proteins or related functional proteins. For example, not only the expression of the STAT3 protein but also the development of the Th17 lineage through the JAK-STAT3 signaling pathway was impaired in AD-HIES.26 Similarly, altered expression of CD132 (IL-2R) or CD127 (IL-7Rα) expression on lymphocytes in SCID, intracellular Wiskott-Aldrich syndrome protein, cytotoxic T-lymphocyte associated protein 4 (CTLA4) in CTLA4 deficiency, and X-linked inhibitor of apoptosis protein in X-linked lymphoproliferative disease type 2 could provide useful information prior to or while waiting for genetic test results.2227
Moreover, FCM could be used for detection of GOF mutations, such as in the STAT1 gene and PIK3CD gene in our cases. FCM tests showed the relative increase of specific proteins (STAT1 and phosphorylated STAT1) or cells (effector memory cytotoxic T cells) compared to healthy individuals in these gene abnormalities, thus with clinical suspicion, they could have suggested the defects in the immune system.2829 Currently, there is no established criteria or cut-off of FCM tests in PIDs although Luk ADW et al.17 has suggested lower than 134/μL based on the CD19+ B-cell counts as an evidence of B cell negative genotypes and elevation of DNT cells expressing αβTCR in peripheral blood has been included in the revised diagnostic criteria for ALPS. More than 1.5% DNT cells out of the total lymphocytes or 2.5% of CD3+ lymphocytes have been proposed as the diagnostic requirement of ALPS; however, it does not provide the diagnosis of specific diseases and elevated DNT can also be observed in other immunodeficient conditions such as CGD and SCID.3031 Therefore, we suggest FCM as one of the screening steps for PIDs.
FCM is being used to monitor treatment. Restoration of a specific immune subset or functional molecules after HSCT can be evidence of successful engraftment.32 Recently, targeted immunotherapy is becoming applicable in some PIDs due to an understanding of the underlying immunopathogenesis. Rapamycin (sirolimus) and mTOR inhibitors for the GOF PIK3CD mutation, and Abatacept, a CTLA-4 agonist, for LOF CTLA-4 or LRBA mutations, were successfully used.3334 FCM may not be helpful during the initial stage due to the unavailability of a specific protein target or non-specific subset pattern; however, it provides evidence for treatment response by normalization of T and B cell differentiation statuses. Thus, in the absence of other objective markers of treatment monitoring, FCM would be helpful.35
Although FCM has been used quite successfully in our center, it has some limitations as a tool for PID diagnosis. First, the genotype and immunophenotype comparability is not complete. The level of protein expression detected by FCM can be dependent on structural variations caused by genetic defects. Less profound structural variation may not affect the protein expression to the level detected by FCM. Second, patients with PIDs commonly demonstrate heterogenous phenotypes, which are not typical for certain genetic abnormalities; therefore, applying a targeted FCM panel selected based on the clinician's first impression may result in the disease being missed. Third, although FCM is less expensive than a genetic test, it could be a burden to apply extensive antibody panels. Fourth, the need of fresh specimens presents another limitation.
To facilitate the diagnosis of PIDs, in addition to an efficient and potent laboratory diagnosis, understanding the current epidemiology of PIDs would be paramount. The number of patients with PIDs in 2013 was estimated to be 6 million worldwide36 and is ever increasing because of the recent progress in laboratory diagnosis. However, the epidemiology of PIDs in Korea has not been well investigated and the frequency is not well known, and only one epidemiological study in Korea used a multicenter questionnaire survey from 2001 to 2005.3 Several countries are currently maintaining their own national registries3738 or participating in multinational networks, which facilitates diagnosis and treatment decisions or manage by enabling information on patients with PIDs to be shared. There is no national registry of PIDs in Korea yet and when considering the continuous development of genetic diagnostic tools, the previous report may not reflect the current situation in Korea. Therefore, establishing a proper registry of Korean PIDs, an optimized network, and referral system for diagnosis and treatment would be expedient in the near future not only for the timely diagnosis to prevent comorbidity and reduce mortality but also for the effective management of the diseases so as to maintain the quality of life.
As described above, many PIDs are complex, and proper diagnosis is critical for improving prognosis. Although FCM might not provide a specific diagnosis, the initial application of FCM, particularly in the spectrum of PIDs with known protein targets on immune cells, would facilitate the timely diagnosis of PIDs, thereby supporting clinical decisions and improving clinical outcome.
 XML Download
XML Download