

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Diabetes is a major public health concern with 3.8 million cases reported in the United Kingdom in 2018.1 There are three major forms of diabetes; type 1, type 2 and gestational diabetes, with type 2 diabetes (T2D) accounting for 90% of cases. T2D is a metabolic disorder whereby the balance between insulin responsiveness of peripheral tissues and pancreatic β-cell insulin secretion is dysregulated. Initial stages of T2D include increased blood glucose and lipid concentrations, with compensatory increase in insulin production. Subsequently, there may be further increases in insulin secretion or advancement to β-cell dysfunction, resulting in hypoinsulinaemia.
Diabetics are at risk of vascular complications, nephropathy, neuropathy, retinopathy, and cardiovascular disease (CVD).2 The leading cause of death in T2D patients is cardiovascular complications, including heart failure (HF) and myocardial infarction (MI).3 In addition, changes in heart function can occur in the absence of hypertension and coronary artery disease, this is known as diabetic cardiomyopathy (DCM). An emerging hypothesis for the mechanism underlying cardiac dysfunction in T2D is altered metabolism in the heart.4
This review focuses on metabolism in the heart and specific changes that occur in T2D, as well as evidence for mechanisms underlying this shift. There will be emphasis on the consequences accompanying these metabolic changes and current efforts to target this. We will consider current therapies, as well as recent developments to target metabolism, which may provide an alternative angle for drug development to pave the way for improved DCM treatment.
CARDIAC METABOLISM IN THE HEALTHY AND DIABETIC HEART
The average heart beats around 100,000 times a day, resulting in a high metabolic demand. This is reflected by the high abundance of mitochondria in cardiomyocytes, as mitochondria are the site of oxidative phosphorylation, the process responsible for 90%–95% of cellular adenosine triphosphate (ATP) production. Substrate breakdown converges on the generation of hydrogen carriers, NADH+ and FADH2 in the mitochondria, which enter the electron transport chain (ETC) and result in ATP production. The remaining 5%–10% of energy production comes predominantly from anaerobic substrate-level phosphorylation in the cytoplasm.
The healthy heart is metabolically flexible and utilises a wide range of substrates for energy production, depending on factors such as oxygen availability, substrate concentration, hormone levels and workload. The most abundantly used substrates by the heart are glucose and fatty acids (FA). Approximately 60% of energy is derived from FA metabolism, 30% from glucose and 10% from other substrates.5 The heart has a limited ability to synthesise FA de novo and depends on long-chain FA transported in the blood as albumin-bound FA, triglyceride-rich very low-density lipoprotein and dietary triglycerides in chylomicrons. In contrast, glucose is freely transported in the circulation.6 Both substrates enter the cardiomyocyte using their respective transporters and are metabolised for the production of ATP (Fig. 1). Glucose is metabolised via glycolysis, whereas FA are metabolised via mitochondrial β-oxidation, prior to convergence of the two pathways at the Krebs cycle.
 | Fig. 1Schematic representing glucose and fatty acid metabolism in the healthy heart. Glucose uptake into cardiomyocytes occurs via GLUTs, namely GLUT1 and GLUT4. Inside the cell, glucose is phosphorylated by HK to G-6-P, which is a central intermediate of metabolism and can enter many pathways. One such pathway is glycolysis, whereby glucose is broken down to pyruvate and a small amount of ATP is generated under anaerobic conditions. Pyruvate can then enter the mitochondria for oxidation or be reduced to lactate in the cytoplasm. Mitochondrial PDH catalyses the oxidative decarboxylation of pyruvate to acetyl-CoA, which can then enter the Krebs cycle to generate hydrogen carriers. In the case of FA, uptake across the sarcolemma occurs primarily by the transporter fatty acid translocase (FAT/CD36). Once within the cardiomyocyte, FA are esterified to LCFA-CoA, which enters mitochondria via CPT1 for β-oxidation, or is incorporated into the myocardial TAG pool. The Krebs cycle yields hydrogen carriers for ATP production at the electron transport chain.GLUT, glucose transporter; HK, hexokinase; G-6-P, glucose-6-phosphate; ATP, adenosine triphosphate; PDH, pyruvate dehydrogenase; FA, fatty acids; LCFA-CoA, long chain fatty acyl coenzyme A; CPT1, carnitine palmitoyl transferase 1; TAG, triglyceride.
|
Although the most commonly associated phenotypic alteration in T2D is increased blood glucose levels, plasma FA levels are elevated to an even greater degree.7 This changes the supply of substrates to the heart and, alongside altered hormonal input, results in altered metabolism. There is an observed increase in FA utilization primarily due to increased delivery and accumulation of lipids in the heart. In diabetic rodents, increased rates of FA oxidation are observed.8 This has been validated by positron emission tomography studies in humans, which show increased levels of overall fat utilisation and FA oxidation in diabetic patients.9 In T2D, there is increased systemic lipid accumulation, including two-fold higher myocardial triglyceride storage, which is associated with impaired cardiac diastolic function.10,11 In conjunction, glucose utilisation is reduced in diabetic hearts and glycolytic rates are suppressed compared with controls. However, there is still contention over whether insulin-stimulated increases in glucose metabolism occur.4 In humans, upon insulin infusion, net increases in glucose uptake are similar between controls and diabetics.12 Conversely, in T2D rats, basal glucose uptake is equal to controls, yet insulin-stimulated glucose uptake was reduced by 50%.13 These contradictory findings may be due to inter-study differences, such as disease severity, patients vs. animal models, and technique used to measure rates of glycolysis.
MECHANISMS FOR METABOLIC SHIFT
1. Cell signalling pathways
Alterations to cell signalling pathways play a significant role in the metabolic shift in the type 2 diabetic heart. Sir Phillip Randle first described the cellular phenomenon whereby utilisation of glucose inhibits FA oxidation and vice versa, named the Randle cycle.14 This cycle ensures that multiple substrates are not needlessly used, however, it has also been implicated in T2D cardiac pathogenesis. Elevated plasma lipid levels lead to increased FA oxidation, and the products of lipid breakdown suppress glucose metabolism, mainly through inhibition of glycolytic enzymes. Long chain fatty acyl coenzyme As (LCFA-CoAs) has been identified as an allosteric inhibitor of hexokinase (HK), the enzyme responsible for the first stage of glycolysis.15 This suggests that increased FA uptake can directly reduce concentrations of glucose-6-phosphate and subsequently glycolysis. Furthermore, increased FA oxidation leads to Krebs cycle-derived citrate accumulation. Citrate can be exported from the mitochondria to the cytosol to inhibit phosphofructokinase, another key enzyme in the control of glycolysis. Phosphofructokinase is also under tight regulation via other substrates including ATP hence, increased FA oxidation exerts its inhibitory effect by also augmenting ATP-mediated inhibition.16 Thus, accumulation of FA metabolic products directly reduces glycolytic flux.
The occurrence of lipid-induced insulin resistance further downregulates glucose metabolism. Upon stimulation of the insulin receptor (IR), glucose transporter 4 (GLUT4) translocates to the sarcolemma, increasing glucose uptake (Fig. 2). In diabetes, elevated FA storage increases myocardial diacylglycerol (DAG) concentrations, a signalling molecule that activates protein kinase C theta (PKCθ). One target of PKCθ is the IR and its adaptor proteins, insulin receptor substrates 1/2 (IRS1/2). Increased intracellular lipid levels in rat skeletal muscle activate PKCθ, permitting phosphorylation of inhibitory serine residues on IRS1. This prevents insulin-induced signalling, reducing GLUT4 recruitment to the membrane.17 In humans, cytosolic DAG content has been correlated to PKCθ activity, and both were increased in T2D compared with control.18 This mechanism needs to be assessed in the heart to see if it is conserved, as currently data is mainly available from skeletal muscle.19
 | Fig. 2Cellular mechanisms that favour FA use within the diabetic cardiomyocyte. Intermediates from FA breakdown inhibit components of glucose metabolism. LCFA-CoA can inhibit HK, the primary enzyme involved in glucose breakdown. Acetyl-CoA from increased FA oxidation can also activate PDK, the inhibitor of PDH. This subsequently reduces PDH activity, reducing pyruvate metabolism. Citrate from the Krebs cycle generated by increased FA metabolism can also inhibit PFK in glycolysis. Overall, this reduces glycolytic flux in the cardiomyocyte. Increased LCFA-CoA leads to increased DAG accumulation, which contributes to altered signalling and increased storage of fats as triglycerides. DAG can activate PKCθ, which has been suggested as the enzyme driving lipid-induced insulin resistance. It has been proposed that PKCθ can phosphorylate serine residues on the insulin receptor and its adaptor protein, IRS1/2. This prevents tyrosine phosphorylation, which is necessary for signalling, reducing translocation of vesicles containing GLUT4 to the membrane, reducing insulin-stimulated glucose uptake.FA, fatty acids; LCFA-CoA, long chain fatty acyl coenzyme A; HK, hexokinase; PDK, pyruvate dehydrogenase kinase; PDH, pyruvate dehydrogenase; PFK, phosphofructokinase; DAG, diacylglycerol; PKCθ, protein kinase C theta; IRS1/2, insulin receptor substrates 1/2; GLUT, glucose transporter; G-6-P, glucose-6-phosphate.
|
2. Transcriptional regulation
Changes to gene expression also contribute to altered metabolism. FA and LCFA-CoA have been reported to act as ligands of at least four transcription factors (TFs): peroxisome proliferator-activated receptor (PPAR), liver X receptor (LXR), and sterol regulatory element binding protein (SREBP), all of which are expressed systemically and, hepatic nuclear factor 4 which is an enterohepatic receptor.20,21 LXRs are nuclear receptors that are activated by oxysterols and LCFAs, and play an important role in controlling glucose, FA and cholesterol metabolism as well as inflammation. Interestingly, they have been reported to be potentially cardioprotective, as well as protective against diabetes due to the effects on glucose metabolism.22 In the liver, LXR activation has been reported to reduce gluconeogenesis and promote increased glucose utilisation. However, activation of these receptors also produces unfavourable lipogenic effects through increasing circulating levels of triglycerides, worsening the metabolic shift in diabetes.23 Similarly to LXRs, SREBPs promote increases in transcription of genes related to fatty acid, triglyceride and cholesterol synthesis, with different isoforms targeting specific genes. SREBP-1c has been reported to upregulate transcription of key enzymes for FA synthesis including acetyl-CoA carboxylase and FA synthase.24,25 Therefore, FA-mediated activation of these TFs can promote upregulation of machinery for further FA and triglyceride production.
PPARs are nuclear receptors with a well-characterised role in control of glucose and lipid metabolism. These receptors are bound by lipids, which act as ligands to the receptor. Upon binding, PPARs can heterodimerise with retinoic acid receptor and then bind to PPAR response element, a specific DNA sequence recognised by PPARs. This binding can then initiate transcription of specific genes that are regulated by these TFs. There are 3 isoforms; PPARα, -β, and -γ, with PPARα being the primary cardiac transcriptional regulator of lipid homeostasis and the isoform most strongly implicated in DCM pathogenesis.26 PPARα exerts control over FA uptake, metabolism and storage through upregulation of genes coding for key proteins involved in these processes. This includes upregulation of fatty acid translocase (FAT/CD36) and fatty acid transport protein, β-oxidation enzymes and carnitine palmitoyl transferase 1 (CPT1). Additionally, PPARα promotes upregulation of pyruvate dehydrogenase kinase 4 (PDK4), the enzyme that inhibits pyruvate dehydrogenase (PDH), hence reducing glucose metabolism (Fig. 3).27 Therefore, increases in FA uptake in T2D promotes activation of lipid-sensitive TFs, enhancing transcription of machinery used for FA metabolism. This further upregulates FA oxidation and suppresses glucose use. This has been validated with animal models, for example, PPARα knockout (KO) mice showed reduced FA and increased glucose oxidation, and were resistant to DCM development.28 Conversely, in hearts of diabetic mice that overexpress PPARα, higher myocardial triglyceride levels alongside a more severe DCM phenotype were observed.29
 | Fig. 3Schematic representing changes mediated by PPARα, which is upregulated in the diabetic heart. Upon FA binding, PPARα becomes activated and dimerises with the retinoic acid receptor. This heterodimer can then bind to the PPAR response element and activate a plethora of genes. This includes genes involved in FA uptake, mitochondrial FA uptake and β-oxidation, including fatty acid translocase (FAT/CD36) and CPT1. Furthermore, PPARα also promotes upregulation of PDK, inhibiting PDH and reducing glycolytic flux.PPAR, peroxisome proliferator-activated receptor; FA, fatty acids; CPT1, carnitine palmitoyl transferase 1; PDK, pyruvate dehydrogenase kinase; PDH, pyruvate dehydrogenase; HK, hexokinase; G-6-P, glucose-6-phosphate; ATP, adenosine triphosphate; LCFA-CoA, long chain fatty acyl coenzyme A.
|
3. Post-translational modifications
More recently, post-translational modifications (PTMs) in cardiomyocytes have also been implicated in T2D pathology. Particularly, changes in lysine acetylation of mitochondrial proteins have been associated with the diabetic heart.30 Acetylation is a PTM involving the transfer of an acetyl moiety from acetyl-CoA onto the ε-amino group of lysine residues. This neutralises the positive charge of the lysine and significantly alters electrostatic properties of proteins.31 This process is highly reversible and is regulated by the activity of acetyltransferases and deacetylases, as well as by the concentration of acetyl-CoA and NAD+.
Although initially identified to occur on histones, a plethora of non-histone proteins are acetylated, including mitochondrial proteins. A large proteomic study by Kim et al.32 showed 277 lysine acetylation sites on 133 mitochondrial proteins. Acetylation of proteins encoded by mitochondrial DNA has also been observed.32 Deacetylation is potentiated by histone deacetylases (HDACs), of which there are multiple different classes. Sirtuins (SIRTs) are class III HDACs that are NAD+-dependent, with three mitochondrial isoforms, SIRT3, 4 and 5 identified (Fig. 4). Studies have suggested SIRT3 is the main deacetylase in cardiac mitochondria.
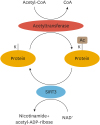 | Fig. 4Schematic representing acetylation and deacetylation of mitochondrial proteins. Acetyltransferases mediate the transfer of an acetyl moiety from acetyl CoA onto lysine (K) residues of proteins in the mitochondria. SIRTs mediate deacetylation, which requires NAD+ as a cofactor, and removes the acetyl group from lysine residues. SIRT3 displays the most robust deacetylating capacity of the mitochondrial SIRTs.CoA, coenzyme A; SIRTs, sirtuins; Ac, acetylation.
|
The majority of acetylation targets in the mitochondria are enzymes involved in the regulation of metabolism.33 Both proteomic analysis of acetylation sites and elucidation of SIRT3 targets have highlighted that enzymes involved in glucose and FA oxidation, the Krebs cycle and oxidative phosphorylation are subject to changes in acetylation status.30 This has led to a surge in studies assessing how acetylation modifies function of different mitochondrial enzymes. The primary mitochondrial enzyme involved in glucose oxidation is PDH. Studies in the liver and skeletal muscle have shown that acetylation reduces PDH activity, and similar observations have since been made in cardiac tissue.34,35 This suggests that increased acetylation reduces metabolism and ATP production from glucose. Assessment of the effects of acetylation on FA oxidation enzymes is less clear. Numerous studies in the liver have shown that acetylation of long-chain acyl co-enzyme A dehydrogenase (LCAD) reduces its function. However, in the heart, hyperacetylation of LCAD and β-hydroxyacyl-CoA dehydrogenase has been observed to increase FA oxidation.36 Furthermore, deletion of cardiac SIRT3 increased acetylation of these enzymes and FA oxidation.36 Studies have also assessed acetylation on Krebs cycle enzymes, but they are limited and contradictory, with findings suggesting opposing effects of acetylation on the activity of a single enzyme.33,34 It is important to consider differences between studies, such as the tissue from which the mitochondria are isolated, differences between in vitro and in vivo acetylation, and how acetylation is mediated, which may account for the variation in results.
In T2D, cardiac mitochondria become hyperacetylated, though the mechanisms underpinning increased acetylation state in T2D are yet to be fully elucidated. There remains contention over whether hyperacetylation is driven by upregulation of acetylating enzymes, or spontaneous acetylation reactions due to high concentration of acetyl-CoA in mitochondria. Acetyl-CoA acts as the donor of the acetyl group which is transferred onto the lysine residue, thus the increased concentrations of acetyl-CoA in the diabetic heart may promote increased mitochondrial protein acetylation levels. Studies have highlighted specific acetylating enzymes; in cancer cell lines, mitochondrial ACAT1 was shown to directly acetylate and inhibit pyruvate dehydrogenase phosphatase 1 (PDP1), resulting in reduced activity of PDH and glycolytic flux.37 However, the lack of acetylating enzymes identified in the heart suggests that non-specific acetylation may be the primary mechanism. The increased accumulation of acetyl-CoA in mitochondria of diabetic hearts, due to increases in FA metabolism, may account for increased acetylation levels.38
Conversely, NAD+ is a co-factor for SIRT3 deacetylation, and so decreased free NAD+ concentrations, as have been reported in the T2D heart, could decrease SIRT3 activity and level of deacetylation. Similarly, SIRT3 may itself be downregulated, reducing the ability of mitochondria to remove acetyl groups from lysine-residues. Studies have shown that in skeletal muscle, Sirt3 expression is downregulated in both type 1 and type 2 diabetic mice models.39 However, data from cardiac tissue is still lacking. Therefore, further research into how changes in acetylation state are mediated is warranted.
CONSEQUENCES OF METABOLIC CHANGES
1. Mitochondrial dysfunction
A primary consequence of altered metabolism is mitochondrial dysfunction. In human skeletal muscle from diabetic patients, reduced ATP synthesis and impaired activity of mitochondrial enzymes have been observed, alongside reduced mitochondrial size and number.35 In T2D models, cardiac mitochondria have reduced ATP production and increased reactive oxygen species (ROS) production compared with controls.40,41 Rodent models have also highlighted differences between mitochondrial populations; subsarcolemmal mitochondria had reduced respiration rates and ETC activity, whereas interfibrillar mitochondria did not.42
Elevated ROS levels are highly implicated in cardiac dysfunction, as they can cause mitochondrial DNA and cellular damage, either via the oxidation of proteins or the generation of toxic lipid peroxidation products.43 Evidence also suggests that mitochondrial permeability transition pores of diabetic cardiac mitochondria have increased propensity to open, allowing release of matrix metalloproteases (MMPs), which can induce apoptosis and increase susceptibility to cardiomyocyte injury.44 ROS may also activate MMPs directly, stimulating myocyte hypertrophy, apoptosis and interstitial cardiac fibrosis.45
In diabetes, baseline oxygen consumption is high due to the dominance of FA utilisation and mitochondrial uncoupling.46 Normally, ATP production is directly coupled with oxygen consumption. However, in dysfunctional mitochondria, uncoupling occurs whereby the proton gradient dissipates, reducing efficiency of ATP synthesis and increasing oxygen use.47 Furthermore, PPARα in the heart may directly upregulate mitochondrial uncoupling protein (UCP) 3. Therefore, the increased FA uptake that occurs in diabetes may drive uncoupling, contributing to reduced ATP synthesis efficiency. Increased expression of UCP3 is associated with increased oxygen use and reduced cardiac efficiency in dietary models of obesity.48
The detrimental processes activated by mitochondrial dysfunction occur in addition to damage by lipotoxic products from the accumulation of triglycerides. For example, ceramide build-up has been shown to stimulate hypertrophy and cardiac dysfunction.49 Lipotoxic intermediates can alter cellular signalling to interfere with ATP production, myocyte contractility and apoptosis.50
2. Cardiac dysfunction
Impaired cardiac energetics, increased oxygen consumption and reduced cardiac efficiency are associated with contractile dysfunction in T2D.51 Healthy hearts are able to maintain the ratio between phosphocreatine (PCr) and ATP (PCr/ATP) in response to increased workload, demonstrating metabolic flexibility. In contrast, diabetic patients have reduced PCr/ATP ratio at baseline, which is further lowered in response to exercise, highlighting the inability to compensate for increased energetic demand.10 This contributes to poor cardiovascular outcomes, as reduced PCr/ATP correlates with increased mortality.52
Structural and functional cardiac changes are also associated with metabolic dysfunction in T2D. Reduced PCr/ATP ratios in diabetic patients correlate with diastolic dysfunction, suggesting that impaired energetics are related to impaired relaxation of the heart.53 Structural remodelling has also been assessed, with increases in concentric left ventricular (LV) hypertrophy alongside small decreases in systolic function observed in people with diabetes.10 This is physiologically relevant as pathological hypertrophy is a key driver of HF. Importantly, these changes were not correlated with alterations in blood pressure, suggesting hypertension was not driving this hypertrophic effect. Furthermore, associations were observed between the level of LV remodeling and myocardial triglyceride storage, with a negative correlation between diastolic function and myocardial triglyceride content reported.11 Hence, metabolic changes may contribute to these structural and functional alterations in the heart, which can negatively affect patient outcome.
CURRENT TREATMENTS FOR DIABETES
The cellular consequences outlined here translate clinically into increased CVD and mortality in patients with T2D. Currently, treatment strategies for T2D include lifestyle interventions, therapeutic agents and exogenous insulin administration. Some patients are able to manage T2D through lifestyle modifications including weight loss and physical exercise. However, the vast majority require therapeutic intervention.2 Drug treatments for diabetes aim to control blood glucose levels, yet studies have shown tight glycaemic control does not decrease the cardiac complications of T2D.54 Independent of treatment status, T2D patients have increased risk for all-cause and cardiovascular-related mortality, as well as HF-related hospitalisation compared with non-diabetics.55
1. Metformin
The first-line treatment for diabetes is metformin. Despite the widespread use of this therapeutic agent, the complex mechanism of action is not fully understood. Classically, metformin is thought to act in the liver to reduce gluconeogenesis, although more recent evidence has indicated activity at other sites, especially the gut. The primary molecular mechanism involves adenosine monophosphate-activated protein kinase (AMPK), which may be activated by metformin via a number of mechanisms. AMPK-independent mechanisms have also been reported, highlighting the complexity of this therapeutic agent.56 Despite the uncertainty in the mechanism of action of metformin, it displays cardioprotective effects, reducing all-cause mortality and MI rates in diabetic patients in RCTs and observational studies.57-59 Furthermore, a study in patients with T2D reported an improvement in LV diastolic function with metformin compared with no treatment.60 This cardioprotective effect may be related to the ability of metformin to rebalance glucose metabolism in cardiomyocytes, via AMPK activation. AMPK has been shown to activate PFK, increasing glycolytic flux. Furthermore, increased AMPK activity has been reported to increase translocation of GLUT4 to the membrane, increasing glucose uptake.61 In vitro studies using showed cardiomyocytes incubated with metformin showed increased AMPK activity as well as increased levels of glucose uptake.62 Overall, metformin may provide beneficial effects by restoring the metabolic shift in cardiomyocytes, which translates clinically into improved cardiac function in diabetic patients.
2. Sodium-glucose co-transporter 2 inhibitors
Sodium-glucose co-transporter 2 (SGLT2) inhibitors are another therapy for T2D. They act through inhibition of renal SGLT2, increasing urinary glucose excretion and lowering blood glucose levels. They have been reported to exert cardioprotective effects, especially reducing incidence of HF,2 however, mechanisms are not fully understood. Three major trials have been conducted assessing the impact of SLGT2 administration on cardiovascular outcomes in diabetes. The empagliflozin (EMPA-REG) outcome and canagliflozin (CANVAS) trials assessed effects in T2D patients with CVD. Empagliflozin reduced risk of cardiovascular-related death and HF in the treatment group, however, no significant effects on atherosclerotic events, namely non-fatal MI and stroke, were observed.63 Similarly, canagliflozin reduced rates of major adverse cardiovascular events (MACE; cardiovascular-related death, non-fatal MI, and non-fatal stroke) and HF.64 Interestingly, the components of MACE were not lowered significantly when considered individually. The Dapagliflozin Effect on Cardiovascular Events-Thrombolysis in Myocardial Infarction (DECLARE-TIMI) trial assessed dapagliflozin in patients with atherosclerotic CVD.65 Dapagliflozin did not reduce rates of MACE compared with placebo, however, incidence of cardiovascular-related deaths and HF-hospitalisation were lowered. Taken together, SGLT2 inhibitors may provide benefit in T2D patients by reducing risk of HF.
Multiple hypotheses on how SGLT2 inhibitors affect the heart have been proposed. One notion is that increased excretion of glucose results in increased ketone body production, which is then utilised in the heart as an alternative fuel to FA. A study in a pig model of HF showed empagliflozin increased myocardial ketone body metabolism and consequentially improved LV ejection fraction.66 Subsequently, the same group showed cardiac function was improved with direct infusion of β-hydroxybutyrate.67 Multiple other mechanisms have been implicated, including stimulation of the AMPK pathway, reminiscent of the mechanism of action of metformin.68 Despite the need for greater research into how SGLT2 inhibitors exert cardioprotective effects, data from large clinical trials suggests they are beneficial in reducing risk of HF, improving outcomes for diabetic patients.
3. Glucagon-like peptide 1 receptor agonists
Glucagon-like peptide-1 (GLP-1) receptor agonists, such as liraglutide and semaglutide, are now well-established treatment options. GLP-1 is an incretin which potentiates β-cell insulin release and inhibits glucagon release hence aiding in glycaemic control. GLP-1 receptors occur at high density in the heart. These agents exert pleiotropic effects in the cardiovascular system including beneficial effects on cardiac metabolism.69 GLP-1 mimetics can enhance glucose uptake and limit FA utilisation, alongside promoting anti-apoptotic and antifibrotic effects in models of T2D.70 Studies in T2D patients have also shown they reduce circulating levels of total low-density lipoprotein-cholesterol, and triglycerides, improving the substrate profile delivered to the heart.71 However, the significance of these effects remains controversial when considering overall cardiac function. In a dog model of DCM, recombinant GLP-1 administration improved LV function.72 Several large clinical trials in human patients have been conducted to elucidate the effects of GLP-1 mimetics. The liraglutide (LEADER) and semaglutide (SUSTAIN-6) trials showed significant reductions in cardiovascular-related deaths and improvements in other secondary outcomes such as non-fatal MI.73,74 Another large trial demonstrated non-inferiority of the once-weekly exenatide treatment compared with placebo when assessing MACE. However, exenatide was not superior to placebo with regard to efficacy.75 Furthermore, trials in patients with advanced HF have shown GLP-1 mimetics exert no beneficial effects.76 The evidence surrounding use of GLP-1 mimetics is still debated with heterogeneity between trials recognised as an obstacle in drawing firm conclusions. However, GLP-1 mimetics are indicated for T2D patients with high risk of cardiovascular events. It is hypothesised they may provide most benefit in reducing atherosclerotic-related events due to their effects on dyslipidaemia.77
4. Thiazolidinediones
Agents such as pioglitazone and rosiglitazone are classified as thiazolidinediones and act to potentiate insulin sensitivity in several tissues; the liver, adipocytes and both skeletal and cardiac muscle. They also act directly on pancreatic β-cells to improve insulin secretion.78 There have been a number of mechanisms proposed, the most well-characterised of which is activation of PPARγ and PPARγ coactivator (PGC1). PPARγ is primarily expressed in adipose tissue and expression is low in tissues that highly express PPARα, including the heart. Similar to PPARα, PPARγ is a TF that controls expression of a large number of genes. One key function is the control of adipocyte proliferation and differentiation, alongside FA uptake. Thiazolidinediones therefore promote increased FA uptake and storage in subcutaneous adipose tissue, reducing levels of circulating FA and triglycerides.79 Thiazolidinediones have also been shown to stimulate specific proteins involved in glucose metabolism, including pyruvate dehydrogenase.2 Overall, by reducing FA delivery to the heart and increasing flux through glycolysis, thiazolidinediones may help to rebalance the metabolic alterations in the diabetic heart. In the UK, thiazolidinediones are contraindicated for people with T2D with HF. The PROactive study demonstrated increased risk of HF leading to hospitalization,80 but reduced risk of MACE compared with placebo. This has also been highlighted in a more recent meta-analysis.81
5. Other therapeutic agents
Despite the potential of the aforementioned therapeutic agents in reducing cardiac complications, other currently used drugs may not provide cardioprotective benefits. Alongside metformin, sulfonylureas are one of the most commonly prescribed antidiabetic agents worldwide. They increase insulin release from pancreatic β cells, reducing plasma glucose levels. However, multiple studies have suggested they may negatively impact cardiovascular function. A number of studies have concluded that sulfonylureas may increase risk of mortality and specific cardiovascular outcomes, including MI, stroke and HF.82-84 Dipeptidyl peptidase (DPP)-4 inhibitors are another class of drugs that have been negatively associated with cardiac outcomes. They act through inhibition of DPP-4, the enzyme that catalyses breakdown of GLP-1. The Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus-Thrombolysis in Myocardial Infarction (SAVOR-TIMI) 53 trial assessed the impact of saxagliptin on cardiovascular outcomes and reported an increase in rates of hospitalisation for HF with no impact on ischaemic events.85 Further clinical trials have been conducted, reporting no increased risk of HF or other cardiac outcomes,86 however, these trials still report no improvement. Overall, the evidence of cardiovascular benefit for most drug treatments in T2D patients with risk of cardiac complications is controversial. This highlights the lack of focus on reducing cardiovascular events in T2D, despite its clear involvement in patient mortality and morbidity. There is a need to focus on specific therapeutics that may improve cardiac function specifically, and that may be used adjunctively to classical glucose-lowering or insulin sensitising agents.
TARGETING METABOLISM FOR THERAPEUTIC BENEFIT
Considering that metabolic changes are clearly implicated in the potentiation of cardiac dysfunction, one avenue for investigation is to target cardiac metabolism directly.
1. Increasing glucose metabolism
A potential mechanism for controlling diabetes-associated cardiovascular comorbidities is to promote glucose metabolism and, via the Randle cycle, reduce FA use. Pre-clinical studies have provided proof of concept that increasing glucose utilisation may be cardioprotective. Ex vivo studies have shown that increasing glucose metabolism with dichloroacetate, a pyruvate mimetic that activates PDH, increases glycolytic flux and improves LV function.87 This has been followed up in vivo, where dichloroacetate administration returned rates of glucose metabolism of diabetic rats to control levels.88 This occurred alongside reversal of impaired diastolic function and normalisation of blood glucose concentrations.
2. Reducing FA metabolism
An alternative mechanism is to reduce FA metabolism directly. Preclinical studies have assessed effects of sulfo-N-succinimidyl oleate (SSO) on cardiac function. SSO is an inhibitor of FAT/CD36 and hence directly blocks FA uptake.89 In diabetic rodents, SSO infusion reduced FA oxidation and cardiac lipid storage back to control levels. Also, cardiac function post-ischaemia was significantly improved compared with untreated rodents. This suggests that reducing FA uptake can reduce FA oxidation and lipotoxicity, improving cardiac function.90 Alternative approaches, such as reducing mitochondrial FA uptake or FA oxidation, via CPT1 inhibitors and β oxidation enzyme inhibitors, respectively, show similar outcomes on metabolism.91 However, reports have suggested that long-term inhibition of CPT1 may mediate deleterious effects on myocardial lipid storage, suggesting that further characterisation may be necessary.92 Furthermore, data has indicated that the anti-anginal drug ranolazine may rebalance metabolism in T2D,93 with trials showing improvements in cardiac function in a diabetic sub-group, highlighting the potential for the repurposing of this drug.94
3. Reducing acetylation
Mitochondrial protein hyperacetylation is one mechanism that has emerged as being involved in the metabolic shift observed in T2D. Consequentially, an emerging area of research is promoting the deacetylation of proteins in the heart. Inhibitors of acetyltransferases, such as the general control of amino acid synthesis 5-like 1 (GCN5L1), may be an option for reducing acetylation of cardiac mitochondrial proteins. GCN5L1 is increased in obese mice with HF, and contributes to increased acetylation and activity of enzymes involved in FA oxidation.95 Activating the deacetylase enzymes may also have potential benefits. Deacetylation of mitochondrial proteins occurs principally by SIRTs. Multiple studies have shown SIRT3 is downregulated in obese and diabetic rodent hearts, and may contribute to cardiac dysfunction, however, this is not ubiquitous throughout reports.96 Recently, there has been assessment of drugs that directly activate SIRT3, such as honokiol. Pillai et al. reported that honokiol entered mitochondria and increased SIRT3 expression by nearly 2-fold, whilst reducing acetylation of mitochondrial proteins in a dose-dependent manner.97 This study also investigated the cardioprotective effects of honokiol, observing antihypertrophic effects in vitro and in vivo in response to pressure overload. Furthermore, honokiol reduced production of ROS and cardiomyocyte death, which was not observed in SIRT3-KO cells, suggesting these cardioprotective effects were driven by specific activation of SIRT3. In 2017, the same group showed honokiol exerted cardioprotective effects against doxorubicin-induced cardiotoxicity, which is often associated with increased ROS, fragmentation of mitochondria and cell death.98 This provides a foundation for research into whether SIRT3 activators, such as honokiol, would provide therapeutic benefits in T2D by improving cardiac mitochondrial function.
CONCLUSION
The diabetic heart is metabolically inflexible and cardiomyocytes utilise FA to a greater extent compared with the healthy heart. This overreliance on FA metabolism and lack of ability to change substrate when necessary, has been directly linked to mitochondrial dysfunction and structural and functional alterations in the heart. Despite this, therapeutics have not yet been developed to target these dysfunctional pathways. Several therapies currently licensed for treatment of T2D are indicated for use in patients at high risk of CVD, with evidence suggesting they may reduce MACEs. However, the vast majority of previous research has only focused on ensuring safety of glucose-lowering agents with regards to cardiovascular events. When considering that cardiac complications are the leading cause of death in diabetic patients, and glycaemic control does not drastically impact risk of CVD, a shift in the focus of T2D treatment is required. This highlights the need for alternative agents that are specifically targeted to the treatment of cardiac dysfunction in T2D. As the significance of metabolic dysfunction in cardiomyocytes is becoming increasingly clear, further fundamental research into the specific mechanisms underlying the metabolic shift is required. This may allow elucidation of potential therapeutic targets, improving therapeutic strategies for cardiac dysfunction in T2D.
 XML Download
XML Download