

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Aging predisposes animals to age-related disorders,12 which include heart failure, systemic metabolic disorders, and arterial diseases.345 Aging itself is not pathogenic, but rather a physiological and biological process that has links with several undesirable diseases.2 To some extent, the mechanisms of aging share similarities with those of age-related diseases.267 For this reason, it remains crucial to understand the pathologies involved in chronological aging and age-related diseases.28 In an elegant article, López-Otín et al.2 reviewed chronological aging and linked this process with alterations in intercellular communication, instability of the genome, telomere shortening, alterations in epigenetics, deregulated nutrient sensing, loss of proteostasis, mitochondrial dysfunction, stem cell exhaustion, and cellular senescence.2 All these processes disturb homeostasis in organs and at the cellular level, and contribute to reduced physiological activity, making organisms prone to death.2 The mechanisms underlying aging and age-related disorders are complex; however, studies have shown that cellular senescence plays pathogenic roles in these conditions.6910111213141516 Cellular senescence was originally defined as cells showing an irreversible termination of proliferation.17 These cells show flat morphology, in association with an altered genetic expression profile.18 They become positive for p53 or p16Ink4a, and contribute to the initiation and maintenance of sustained chronic sterile inflammation.268121419 Cells positive for these senescence markers accumulate in organs affected by aging and age-related disorders.719 Co-culture of young fibroblasts with senescent fibroblasts increased the levels of markers of DNA damage in young cells, indicating that senescence has a contagious aspect, which was first described as the “bystander effect”.20 In general, senescent cells are considered to promote tissue remodeling.68 It was recently shown that a process referred to as “senolysis” that involves the pharmacological or genetic depletion of senescent cells yields beneficial effects in chronological and age-related disease models.212223 Compounds that mediate cell death only in senescent cells are referred to as “senolytics,” and studies have suggested that the use of senolytics is promising for combating age-related disorders.92324
CELLULAR SENESCENCE AND SENESCENCE-RELATED MOLECULES IN ARTERIAL DISEASES
The number of senescent cells in arteries is reported to increase significantly with age.7252627 Aging is associated with higher expression of cyclin-dependent kinase inhibitors (p16 and p21), phosphorylated p38, double-stranded DNA breaks, and senescence-associated beta-galactosidase (SA-β-gal) activity in the vessels of humans and rodents.28293031 The expression of p53, a tumor suppressor gene, and p21 was found to be increased in the arteries of an aged population.27 This was associated with the breakdown of telomere structures, described as “telomere uncapping”.27 A study analyzing single-nucleotide polymorphisms concluded that elongated telomere length is associated with reduced risks of coronary heart disease and abdominal aortic aneurysm (AAA).32 Additional studies have indicated that cellular senescence, mediated by senescence molecules, plays a role in the pathogenesis of arterial diseases, including aortic aneurysm, coronary artery disease, and peripheral artery disease (PAD), all of which are discussed below.2533
1. Aortic aneurysm
Thoracic aortic aneurysms and AAAs are life-threatening aortic diseases, the risk of which is known to increase with age. Endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) from AAA patients show telomere attrition and oxidative DNA damage.25 Human VSMCs from AAA patients exhibited high levels of phosphorylated histone H2AX (γH2AX), but no correlation was found with patients' chronological age.33 Sirtuins are protein deacetylases that are known to inhibit aging and contribute to longevity.34 Suppression of vascular cell senescence by the activation of sirtuin 1 (SIRT1) prevented AAA formation, but AAA formation was augmented by promoting cell senescence in these cells.35 It is well known that calorie restriction (CR) can extend the lifespan in organisms ranging from Caenorhabditis elegans to mammals.3637 CR can reduce the incidence of AAA formation induced by the peptide hormone angiotensin II (AngII) in mice.38 Systemic genetic depletion of p53 also contributed to the suppression of AAA formation.39 Patients with thoracic aortic aneurysm/dissection exhibited elevated numbers of p53-, p21-, and p19-positive cells in the vessel wall, along with increased SA-β-gal activity.40 These results indicate that cellular senescence plays a pathogenic role in aortic aneurysm.383940
2. Coronary artery diseases
Studies have suggested a close connection between cellular senescence and coronary artery disease.741 In patients with ischemic heart disease, the number of senescent cells was higher in the coronary arteries than in the internal mammary arteries.7 A large amount of evidence now indicates that telomere length and telomerase activity in peripheral leukocytes and circulating progenitor cells are involved in human cardiovascular diseases.4243 It has been reported that endothelial progenitor cells from coronary heart disease patients exhibited telomere attrition and reduced telomerase activity.41 In addition, leukocyte telomere length showed an inverse association with the risk of coronary heart disease independent of conventional vascular risk factors.44 MicroRNA (miRNA) is a type of small non-coding RNA, and studies have indicated that some subsets of miRNA play a role in either maintaining or disturbing systemic homeostasis.4546 Expression of miR-23a has been shown to be higher in peripheral blood mononuclear cells in patients with coronary artery disease than in controls.47 Telomeric repeat binding factor-2 (TRF-2) plays a critical role in telomere maintenance, and miR-23a has been shown to reduce the levels of TRF-2.47 The level of miR-23a was higher in coronary artery disease patients than in controls, suggesting that it affects the progression of coronary atherosclerosis via downregulation of TRF-2.47 Nutrients and oxygen are delivered via capillaries, and the formation of the capillary network is critical for maintaining organ function.1148 Vascular endothelial growth factor-A (VEGF-A), an angiogenic molecule, has been extensively studied, and suppression of VEGF-A has been reported to result in a lack of tissue remodeling.1148 Several splice variants or isoforms of VEGF-A exist, all of which are generally recognized to elicit a pro-angiogenic response; however, some isoforms have been reported to become anti-angiogenic molecules.4950 For example, under metabolic stress, it has been shown that levels of VEGFA165b increased and became pathogenic by reducing angiogenesis in a murine ischemic limb model.50 Senescent human aortic ECs (HAECs) express a high level of VEGFA165b, and interestingly, the transcript Vegfa165b is present at higher levels in patients with coronary heart disease.51 These results indicate that dysregulation of the splicing of VEGF-A into its various isoforms may be a key feature of EC senescence involved in the pathogenesis of coronary heart disease.51
3. PADs
The ankle-brachial index (ABI) is defined as the ratio of ankle systolic blood pressure to that of the arm. A decline in the ABI is known to be indicative of PAD.52 As the ABI decreases with age, this index has traditionally been considered to be independent of traditional risk factors for PAD.53 The prevalence of PAD, and in particular pathological PAD, increases with age.54 It was reported that aging caused a drop in perfusion in a hind limb ischemia model.55 Furthermore, aging was shown to exacerbate injuries mediated by ischemia-reperfusion in skeletal muscle.56 In one study, hind limb ischemia was generated in young, old, and very old mice administered a telomerase activator.57 Blood flow recovery was suppressed in the ischemic limbs of old mice compared to young mice, and recovery in old mice improved in response to administration of a telomerase activator, TA-65.57 The expression of hypoxia inducible factor 1α, VEGF-A, and peroxisome proliferator-activated receptor gamma coactivator 1-alpha was reduced in old mice, but increased following TA-65 administration.57 Expression of senescence markers—including p53, p21, and p16—increased in ischemic limbs on day 3 in old mice, although the expression of these senescence markers was ameliorated in old mice given TA-65.57 This indicates that telomerase activation is a potential therapeutic agent for ischemic tissues in aged individuals.57 Serum from PAD patients showed acceleration in the aging phenotype of HAECs.58 This was suppressed by administering sulodexide (a mixture of glycosaminoglycans).58 The results of that study indicate that serum from PAD patients contains pro-senescent molecules that accelerate arteriosclerosis.58 However, to date, few studies have investigated the role of cellular senescence in PAD or the role of senescence processes in VSMCs specifically.57
ROLE OF SENESCENCE AND SENESCENCE-RELATED MOLECULES IN VSMCs
It is well known that VSMCs, ECs, and immune cells play crucial roles in maintaining vascular homeostasis.5960 These cells are involved in complex cell-cell interactions, but no investigative techniques currently exist for thoroughly determining the role these cells play in vascular structure, function, and remodeling.60 Studies have indicated, though, that senescence processes play a pivotal role in the involvement of these cells in arterial diseases.72561
1. VSMCs
With aging, VSMCs undergo transformation from a “contractile” to a “synthetic” phenotype, show enhanced inducible nitric oxide synthase activity, and exhibit high expression of intercellular adhesion molecule-1 (ICAM-1) and angiotensinogen in response to stress.362 Senescent VSMCs have been reported to accumulate in atherosclerotic plaques of patients with ischemic heart disease, AAA, and PAD.725 Approximately 20% of the VSMCs in a carotid artery plaque stained positive for p16, p21, and SA-β-gal.61
In cell culture conditions, VSMCs from the fibrous cap of an atheroma exhibited telomere attrition compared to those from normal media.61 Furthermore, VSMCs with short telomeres were found to be positive for SA-β-gal activity, in addition to their increased levels of p16 and p21.61 Oxidative stress induced DNA damage in VSMCs, which was associated with a reduction in telomerase activity, telomere attrition, and cellular senescence.61 In atherosclerotic plaques, VSMCs become senescent, and the levels of TRF-2 are reduced in these cells.63 TRF-2 is known to be localized in telomeres, where it mediates their protective activity.64 In vitro studies have demonstrated that increased expression of TRF-2 reduces DNA damage, accelerates DNA repair, and suppresses the process of senescence.65 Inhibition of TRF-2 resulted in the opposite phenotype.65 Finally, VSMC-specific TRF-2 suppression led to the progression of atherosclerosis in vivo in mice, which was ameliorated in VSMC-specific TRF-2 overexpression in vivo in mice.65 The peptide hormone AngII is involved in vasoconstriction, ultimately resulting in increased blood pressure.66 It was previously reported that AngII induced senescence in VSMCs in Apoe−/− mice.67 Smooth muscle 22α, an actin-binding protein, was recently shown to promote AngII-induced senescence by the suppression of E3 ubiquitin ligase (Mdm2)-induced p53 degradation in mice.68 Senescent VSMCs in carotid plaques stained positive for interleukin-6, suggesting that senescent VSMCs contribute to vascular remodeling by acquiring the senescence-associated secretory phenotype.69 Dysregulation of the production of growth factors and modifiers of the extracellular matrix results in senescent VSMCs, further accelerating the remodeling process in vessels.69 Suppression of SIRT1 in VSMCs increased vascular cell senescence, upregulated p21 expression, enhanced vascular inflammation, and ultimately promoted pathologies associated with AAA.35 In contrast, specific SIRT1 overexpression inhibited AngII-induced AAA formation and progression in Apoe−/− mice.35
It is well known that CR can prolong the lifespan.70 Interestingly, CR has also been shown to contribute to vascular health, as CR inhibited AAA formation in AngII-infused Apoe−/− mice.38 Furthermore, in an AAA mice model, CR led to an increase in SIRT1 expression in VSMCs, and genetic depletion of Sirt1 in VSMCs abolished the protective role of CR.38 Nicotinamide phosphoribosyltransferase (NAMPT) is a rate-limiting enzyme that initiates nicotinamide adenine dinucleotide (NAD+) production.71 Reduced NAD+ levels are associated with aging and age-associated diseases and contribute to the progression of pathologies in these disorders.71 In ascending dilated aortas from patients with aortopathy, an inverse relationship was found between NAMPT in VSMCs and aortic diameter.72 Depletion of Nampt in VSMCs led to dilatation of the aorta in association with a 43% reduction in NAD+ in the media.72 Infusion of AngII in a VSMC-Nampt knockout mouse model led to aortic medial hemorrhage and dissection or tearing.72 In addition, VSMCs showed high levels of p16 and SA-β-gal activity, indicating that they were senescent, not apoptotic.72 These results suggest that suppression of VSMC senescence contributes to the inhibition of pathologies associated with atherosclerotic diseases.387172
2. ECs
It is well known that ECs play a role in the maintenance of vascular homeostasis, and that EC dysfunction is associated with a pro-inflammatory phenotype, an altered angiogenic response, and pro-oxidant and pro-thrombotic states.151673 The proliferation and migration capacity of aged ECs is diminished, which is associated with reduced nitric oxide production.74 Studies have suggested that EC dysfunction plays a causal role in alterations of blood pressure, systemic metabolism, and the coagulation system, and may play a pivotal role in cellular senescence.75 In a murine left ventricular pressure overload model, the expression of p53 was increased, leading to overexpression of ICAM-1 in these cells, which promoted cardiac inflammation.16 Levels of p53 in ECs also increased under metabolic stress, ultimately resulting in diminished endothelium-dependent vasodilatation and ischemia-induced angiogenesis.15 In addition, elevated levels of p53 in ECs induced systemic metabolic dysfunction.75 Senescent ECs have been shown to exist in atherosclerotic plaques in coronary arteries.7 In contrast, the expression of these senescent cells was low in the internal mammary arteries.7 In the coronary arteries, SA-β-gal activity was high in cells located on the luminal surface, indicating that ECs in the coronary arteries contain SA-β-gal.7 Disturbance of flow in the ascending aorta and aortic arch was shown to promote EC senescence in atherosclerotic mice.76 Oxidized low density lipoprotein (LDL) is a well-established atherogenic lipoprotein.77 The most electronegative subfraction of LDL is L5, which was recently demonstrated to promote EC senescence.63 Furthermore, L5 administration increased levels of γH2AX and SA-β-gal activity in the aortic endothelium of mice.63 A high-caloric diet increased L5 levels in circulation, which also promoted a senescent-like phenotype in the aortic endothelium of Syrian hamsters.63 Regarding cellular pathways, an in vitro study showed that L5 induced EC senescence via activation of the ATM/Chk2/p53 pathway.63 Long non-coding RNA H19 (lncRNA H19), has been shown to be downregulated in the endothelium of aged mice.78 The loss of lncRNA H19 led to increased p16 and p21 levels in vitro.78 In addition, lncRNA H19 depletion increased expression of signal transducer and activator of transcription 3 (STAT3), while suppression of lncRNA H19 reduced STAT3.78 Additional studies have reported that increased H19 levels contributed to an inhibition of STAT3-mediated cell senescence.78 Several molecules and methods have been reported to inhibit senescence in ECs.7980 Polyphenol suppressed reactive oxygen species (ROS) production, reduced endothelial p53 levels, and increased nitric oxide levels in the aorta, contributing to an inhibition of endothelial dysfunction.79 Both L-citrulline and L-arginine supplementation reduced SA-β-gal activity and p16 levels, and increased telomerase activity in human umbilical vein ECs maintained in a high-glucose medium.80 Administration of these amino acids also led to a reduction of SA-β-gal activity in the abdominal aorta of Zucker diabetic fatty rats with diabetes.80 Omega-3 fatty acids (eicosapentaenoic acid and docosahexaenoic acid) suppressed a hydrogen peroxide-induced increase in SA-β-gal activity in HAECs.81 Aging increased levels of the senescence marker p19 in arteries, and this was suppressed by dietary rapamycin administration, resulting in enhanced endothelium-dependent dilatation in carotid arteries.82 In humans and rodents, it is well known that physical activity improves vascular structure and function.83 The bioavailability of nitric oxide decreases with age, which is associated with high ROS levels and reduced physical activity.84858687 Compared to sedentary older individuals, older individuals who regularly exercised had lower levels of p53, p21, and p16 in ECs from the brachial artery and antecubital vein.88 This indicates that physical activity inhibits cellular senescence in human vessels.88
3. Immune cells
Atherosclerotic lesions develop and progress through excessive levels of lipids and chronic inflammatory responses.60 Evidence indicates that cellular senescence in immune cells plays a causal role in the progression of atherosclerosis.23899091 Links between immune cell senescence and atherosclerosis have been reported.23899091Telomere attrition in leukocytes in aged individuals has been linked with a high mortality rate, of which a moderate number of cases are attributable to cardiovascular deaths.23899091 Monocytes from atherosclerotic patients showed high levels of ROS and pro-inflammatory cytokine production.89 Senescent intimal foam cells have been shown to accumulate in atherosclerotic lesions and become drivers of atheroma formation.23 Another report showed that p16INK4a can induce cellular senescence in macrophages, causing these cells to become pro-inflammatory.91 Evidence indicates that leukocyte senescence can accelerate plaque formation in atherosclerosis.2391 Inhibition of the senescence process in immune cells is another important concept to be considered in the treatment of atherosclerotic vascular disorders.238991
SENOLYTICS TARGETING ARTERIAL DISEASES
Genetic and pharmacological studies have indicated that specifically eliminating senescent cells may contribute to reversal of the aging phenotype.992 A study in INK-ATTAC mice (prematurely aging mice) eliminated p16Ink4a-positive cells by introducing AP20187, thereby reversing the aging phenotype in several organs including white adipose tissue, the heart, and the kidney.9 Elimination of p16Ink4a-positive cells led to a significant reduction in cardiomyocyte cross-sectional area, indicating an increase in cardiomyocyte number.22 Currently, more than 10 compounds have been reported to induce senolysis, the specific depletion of senescent cells.73 Currently, dasatinib and quercetin (D+Q) and ABT263 are the most studied senolytics.247392 In particular, D+Q has been tested in clinical studies targeting chronic kidney diseases or idiopathic pulmonary fibrosis.92 Recently, it was reported that D+Q administration reduced p16INK4a, p21, and SA-β-gal levels in subcutaneous adipose tissue.92 This was associated with the suppression of inflammatory cell infiltration and an increase in the number of adipocyte progenitor cells, indicating that senolytics may be beneficial in humans.92 In vitro studies of ABT263 showed that this compound specifically depleted irradiated fibroblasts, and that administration of ABT263 led to the clearance of p16Ink4a-positive cells in bone marrow.24 Importantly, ABT263 was shown to contribute to the rejuvenation of hematopoietic stem cells during aging.24 In an aged murine model with hypercholesterolemia, D+Q treatment yielded improvements in vasomotor function.93 In 24-month-old mice, D+Q introduction ameliorated systolic cardiac dysfunction and inhibited end-systolic left ventricular dilatation.94 In pharmacological and genetic senolytic models, the depletion of senescent cells in aged mice contributed to the activation of cardiac progenitor cells in the heart, along with an enhanced proliferative capacity of cardiomyocytes.2195 Evidence indicates that senolytics have the potential to be considered as possible therapies for atherosclerotic vascular disorders.23 In addition, p16Ink4a-3MR (3MR) is another transgenic mouse model generated to study the inducible depletion of p16Ink4a positive cells.23 Specific deletion of p16-positive cells, as analyzed in Ldlr−/−, p16-3MR mice, led to the elimination of macrophages, VSMCs, and ECs expressing p16 in vessels, and reversed atherosclerosis.23 In a hypercholesterolemic mouse model, D+Q administration reduced aortic calcification and osteogenic signaling.93 In an aged murine myocardial infarction model, ABT263 administration improved myocardial remodeling, diastolic function, and overall survival.96 These results suggest that elimination of senescent cells with senolytics should be considered as a next-generation therapy for atherosclerotic disorders.6247392
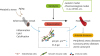
The most fundamental limitation of currently available senolytics is their potential toxicity.9798 Dasatinib is a tyrosine kinase inhibitor and ABT-263 is a Bcl-2 family inhibitor; it is well known that both were developed as anticancer drugs and therefore come with a risk of toxicity.9798 Quercetin is polyphenol in the flavonoid group, and may be less toxic; however, it would, in general, need to be taken in combination with dasatinib.73 Discovering new senolytic compounds continues to be an interesting and important research area to combat arterial diseases (Fig. 1).
Fig. 1
The emerging concept underlying therapies targeting arterial diseases with senolysis. The rejuvenation of senescent vascular cells by senolysis contributes to the suppression of senescence markers such as p53/p21, p16Ink4a, and SA-β-gal, and reverses atherosclerosis. In addition to genetic models, senolysis includes pharmacological models such as the D+Q and ABT263 models. These contribute to the suppression of pathologies in arterial diseases, including peripheral artery disease, coronary artery disease, and aortic aneurysm.
D+Q, dasatinib and quercetin; SA-β-gal, senescence-associated beta-galactosidase.
CONCLUSION
In this review article, the role of cellular senescence in arterial diseases was discussed. In combination, the results from many studies show that senescent cells play a pathogenic role in several vascular diseases, including aortic aneurysm, coronary artery disease, and PAD. Genetic and pharmacological senolytic approaches have made considerable contributions to the regression of atherosclerotic plaques in older patients. Findings from more recent studies have indicated that these 2 approaches should be considered as therapeutic possibilities for aging individuals, as they do not appear to promote tumorigenesis. The concept of senolysis has opened a new and very interesting avenue in aging research. In addition to further research investigating the underlying mechanisms contributing to the progression of cellular senescence, it remains critical to find less toxic senolytics in order to establish next-generation therapies for atherosclerotic vascular disorders.
 XML Download
XML Download