

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The Western lifestyle is characterized by high dietary lipid intake and a relatively limited level of physical exercise, which together often result in the development of obesity. As obesity becomes more severe, excess lipids cannot be stored in adipose tissue, which overflows, and as a consequence lipids are deposited ectopically both in the vessel wall (atherosclerosis) and in non-adipose tissues, such as the liver, heart, and skeletal muscle.12 Ultimately, ectopic lipid storage leads to organ dysfunction. For instance, obesity triggers a marked change in the control of glucose and lipid metabolism in the heart, whereby long-chain fatty acids (LC-FA) become the major substrate for energy provision, while glucose utilization is hampered due to the development of insulin resistance. This change in substrate preference underlies the well-known association of obesity with type 2 diabetes and cardiovascular complications such as acute myocardial infarction and so-called diabetic cardiomyopathy.34 Taken together, these observations indicate that it is a metabolic challenge for the heart to control the rate of fatty acid uptake and utilization at all times, balancing the need to secure sufficient fatty acids for energy provision, thereby avoiding a possible energy deficit, while avoiding an unphysiologically high uptake rate that may lead to deleterious triacylglycerol accumulation and, ultimately, lipotoxic heart failure.
In this review, we summarize the current knowledge on the regulation of myocardial fatty acid utilization. Emerging evidence indicates that cluster of differentiation 36 (CD36; officially designated as scavenger receptor B2 [SR-B2]), a transmembrane glycoprotein, plays a pivotal role both in the healthy heart and in the development of metabolic cardiac diseases, such as diabetic cardiomyopathy. More recent studies have suggested that CD36 may be a suitable target for altering myocardial fatty acid utilization, especially in cases of lipid overload. Furthermore, new knowledge on subcellular proteins that influence CD36 function in a tissue-specific manner may even enable the development of approaches to manipulate fatty acid utilization specifically in the heart.
MYOCARDIAL FATTY ACID UPTAKE
LC-FA are the predominant substrate for energy provision in the heart, and are derived from both circulating lipoproteins and the pool of fatty acids non-covalently bound to plasma albumin.5 In the healthy heart, fatty acids contribute to about 60% of the myocardial ATP production rate, the remainder being provided mostly by glucose (25%–30%) and lactate (10%) with minor contributions from ketone bodies and amino acids (also depending on the dietary state). The amount of substrates stored endogenously in cardiac myocytes (i.e., glycogen and triacylglycerols), suffices to maintain contractile activity for only a limited duration (minutes), indicating that a virtually continuous delivery of exogenous substrates is mandatory for proper cardiac function.
On their way from the plasma to cardiomyocytes, fatty acids must pass through the endothelium, which in the heart—unlike the liver—is characterized by few, small fenestrae that do not allow the rapid passage of larger proteins such as albumin. Therefore, fatty acids are transported not alongside, but across, endothelial cells to reach the interstitium where they are bound to albumin. The concentration of albumin in the interstitial space is about one-third of that found in the plasma. The subsequent uptake of fatty acids into cardiomyocytes is facilitated by membrane-associated proteins, with a predominant role played by the transmembrane glycoprotein CD36, with additional roles played by plasma membrane fatty acid-binding protein (FABPpm) and a number of so-called fatty acid transport proteins (FATPs; FATP1, FATP4, and FATP6).67 CD36 is a member of the superfamily of scavenger receptor proteins - class B, and is officially designated as SR-B2.8 FABPpm is a 40–43 kDa peripheral membrane protein present at the outer leaflet of the sarcolemma that was first described in 19859; however, it has hardly been studied, and its precise physiological function therefore remains elusive. FABPpm likely acts in concert with CD36 to take up fatty acids by facilitating their passage through the unstirred water layer immediately adjacent to the lipid bilayer. FATPs are actually acyl-CoA synthetases that stimulate the rate of cellular fatty acid uptake by converting incoming fatty acids directly into their acyl-CoA thioester, which is collectively referred to as metabolic trapping.10 FATPs are primarily involved in the uptake of very-LC-FA (C22 and beyond), and are less important for the bulk uptake of palmitate and of other abundant long-chain fatty acid species.10
1. Dynamic role of CD36 in myocardial fatty acid uptake
The glycoprotein CD36 is an 88-kDa transmembrane protein that is composed of 2 transmembrane domains and a large extracellular loop containing 9 glycosylation sites and 2 phosphorylation sites.711 Both the C-terminal and N-terminal of the protein are located intracellularly, as part of small cytoplasmic tails. The extracellular loop of CD36 contains a hydrophobic binding pocket that functions as an acceptor of fatty acids to promote the partitioning of fatty acids and their delivery to the outer leaflet of the lipid bilayer. Thereafter, fatty acids ‘flip–flop’ to the inner leaflet of the membrane, a process that occurs very quickly and does not need assistance from membrane proteins.12 The subsequent and final step in the transmembrane transportation process is desorption of fatty acids from the inner leaflet and their binding to the cytoplasmic fatty acid carrier (FABPc). This desorption is regarded as the rate-limiting step of overall transmembrane transport. CD36 is considered to facilitate this step by providing a docking site for FABPc or acyl-CoA synthetases.7 The presence of CD36 largely in specific membrane domains designated ‘lipid rafts’ may indicate the existence of areas in the sarcolemma with a clustering of the specific proteins and enzymes involved in transmembrane fatty acid transport, which would result in a highly efficient uptake process.
Studies of isolated rodent cardiac myocytes during electric field stimulation aimed at increasing contractile activity revealed that the resulting increase in fatty acid uptake rate was accompanied by an increased amount of CD36 in the sarcolemma.1314 The latter increase was due to the translocation of CD36 from intracellular storage depots, most notably endosomes, to the sarcolemma, a process that could be triggered on a time scale of minutes. Furthermore, it was observed that CD36 translocation from endosomes to the sarcolemma could be triggered with similar speed by the hormone insulin,15 as visualized by 2-photon microscopic imaging (Fig. 1). Upon removal of insulin, CD36 is rapidly internalized, leading to a concomitant decrease in the fatty acid uptake rate.16 These changes in the sarcolemmal CD36 presence are due to the translocation and internalization of CD36, as the total cellular amount of CD36 remains unaltered. Subcellular recycling of CD36 between endosomes and the sarcolemma as a mechanism to regulate the rate of fatty acid uptake by cardiac myocytes closely resembles the mechanism by which glucose uptake is regulated by subcellular recycling of the glucose transporter (GLUT) 4, which also is triggered by increased contraction and the presence of insulin.17 Unlike CD36, its obligatory partner FABPpm is not regulated by translocation and is permanently present at the cell surface, implying that FABPpm plays a merely permissive role in fatty acid uptake. Most likely, upon exposure of cells to either electric field stimulation or to insulin, CD36 translocates to a specific area of the sarcolemma (lipid raft), and thereafter binds to FABPpm to form a functional uptake complex. Taken together, upon increased muscle contraction or insulin stimulation, both CD36 and GLUT4 are recruited to the sarcolemma within the same time frame, resulting in increased uptake rates for both fatty acids and glucose (Fig. 2).18
Fig. 1
Two-photon microscopy imaging of sarcolemmal CD36 before and after insulin stimulation. Isolated rat cardiac myocytes under basal conditions (left) or after 15 minutes of incubation with 100 nM insulin (right) were incubated with anti-CD36 antibody and subsequently with a fluorescein isothiocyanate-labeled secondary antibody (green). The more intense green labeling of the sarcolemma (right photo) is due to the insulin-induced translocation of CD36 from intracellular storage depots (endosomes) to the sarcolemma. Courtesy of Dr. L.K.M. Steinbusch.
CD36, cluster of differentiation 36.
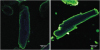
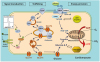
Fig. 2
Scheme illustrating the similarity of the regulation of CD36-mediated LC-FA uptake and GLUT4-mediated glucose uptake by cardiomyocytes. In response to contractile activity or stimulation with insulin, both the fatty acid transporter CD36 (also designated scavenger receptor B2) and the GLUT4 translocate to the sarcolemma to increase fatty acid and glucose uptake, respectively. Note that CD36 and GLUT4 may be recruited from distinct stores within the endosomal compartment. The GLUT1 is constitutively expressed in the sarcolemma and contributes up to 25% of glucose uptake.18
CD36, cluster of differentiation 36; GULT, glucose transporter; LC-FA, long-chain fatty acids; FABPpm, plasma membrane fatty acid-binding protein; FABPc, cytoplasmic fatty acid-binding protein.
The availability of CD36-null mice has made it possible to study the contribution of the CD36-facilitated pathway to the overall rate of myocardial fatty acid uptake. Habets et al.19 showed that under control conditions, palmitate uptake by cardiomyocytes from CD36-null mice was not different from that in cardiomyocytes from wild-type mice due to a compensatory upregulation of FATP1 in the CD36-null mice. However, during electric field stimulation the cardiomyocytes from CD36-null mice failed to upregulate the palmitate uptake rate, while cardiomyocytes from wild-type mice showed an almost 3-fold higher uptake rate. From these data, the authors concluded that in contracting myocytes, CD36 is responsible for about 70% of the rate of fatty acid uptake.19
2. Rate-limiting kinetic step in myocardial fatty acid utilization
Fatty acid import into the mitochondria has been considered to be the major rate-limiting kinetic step in the overall fatty acid utilization process in the heart.520 This step is regulated by carnitine palmitoyl-transferase-I (CPT-I) via its physiological inhibitor malonyl-CoA. However, from a physiological viewpoint, it would make sense to regulate the overall rate of cellular fatty acid utilization at the level of entry of fatty acids into the cardiomyocyte, because doing so would avert the risk of overloading the cell with fatty acids that cannot be immediately oxidized (posing a risk of lipotoxicity). Indeed, evidence is increasing that cellular fatty acid uptake, not CTP-I, is the rate-limiting step.21 Specifically, a study of rats administered the pharmacological compound etomoxir, a direct inhibitor of CPT-I, showed that inhibition of CPT-I activity by about 50% had no effect on cardiac fatty acid utilization as measured in vitro in cardiomyocytes isolated from these rats, neither with low nor high external fatty acid concentrations and neither in quiescent cells nor during electric field stimulation of the cells.22 As a result, it can be concluded that CPT-I mostly plays a permissive role in overall myocardial fatty acid utilization, while sarcolemmal fatty acid transport is rate-governing.
LIPID-INDUCED CARDIAC DYSFUNCTION
1. Key role of CD36 in the pathogenesis of diabetic cardiomyopathy
A chronically elevated fatty acid supply to the heart, as occurs when a high-fat diet is consumed and in obesity, is known to induce a shift in myocardial energy provision towards an increased utilization of fatty acids at the expense of glucose.23 Such a substrate switch eventually may lead to the accumulation of specific lipid species in cardiomyocytes together with mitochondrial dysfunction, subsequently resulting in insulin resistance. In turn, insulin resistance is a causal factor for type 2 diabetes with impaired cardiac contractile function, which is referred to as diabetic cardiomyopathy.3 The course of events that eventually result in diabetic cardiomyopathy has been elucidated in the last decade, and evidence indicates that CD36 plays an early pivotal role.
Chronic oversupply of fatty acids to the heart triggers alterations in the subcellular recycling of CD36. In the normal heart, the total cellular amount of CD36 is distributed roughly evenly between endosomes and the sarcolemma, but following chronic lipid oversupply, CD36 is mostly found at the sarcolemma at the expense of endosomal CD36 (i.e., there is a net relocation of CD36 from endosomes to the sarcolemma). After starting a high-fat diet, this subcellular redistribution occurs very rapidly (e.g., within 2–3 days in the skeletal muscle of rodents),24 and is accompanied by a concomitant increase in the rate of fatty acid uptake before any other change in muscular metabolism. As a result, over the course of about 2 weeks, fatty acids become the main metabolic substrate for energy provision, while the use of glucose is downregulated.24 However, the increase in fatty acid uptake is larger than needed by the myocytes for metabolic energy provision, so that the intracellular storage of fatty acids as triacylglycerols markedly increases. This pattern of increased myocardial lipid formation has been observed previously,2 and was proposed to be related to the limited ability of adipose tissue to accommodate the overwhelming plasma availability of fatty acids, as a result of which fatty acids are stored ectopically in the heart and liver (myocardial and hepatic steatosis). An increased amount of intracellular triacylglycerols is generally accompanied by increased levels of fatty acid metabolites, especially diacylglycerols and ceramides, 2 compounds that are known to inhibit insulin signaling, thereby causing insulin resistance.25 The main consequence of insulin resistance is a lowered glucose uptake rate due to impaired translocation of GLUT4 from endosomes to the sarcolemma. In such conditions of chronic lipid overload, the heart relies predominantly on fatty acids for metabolic energy provision because it cannot take up sufficient amounts of glucose, and it shows marked contractile dysfunction.26
This cascade of events was mimicked in isolated rat cardiomyocytes by subjecting the cells to a culture medium with a high concentration of palmitate (e.g., 200 µM). After 48 hours in culture, the cardiomyocytes showed a 2-fold higher presence of CD36 in the sarcolemma, almost 3-fold higher intracellular triacylglycerol content, insulin resistance (measured as the response of Akt phosphorylation to exogenous insulin), and a peak sarcomere shortening reduced to approximately 50% of that found in cardiomyocytes cultured in a low-palmitate medium (20 µM palmitate) (Fig. 3).27 Furthermore, the pivotal early role of CD36 in this cascade is evidenced by the fact that addition of anti-CD36 antibodies to the culture medium completely prevented changes in each of these parameters and, importantly, prevented the loss of cardiac contractile function (Fig. 3). These in vitro findings are in line with published reports on mice in vivo, as CD36-null mice were found to be protected against high-fat diet-induced loss of cardiac function.2829 Together, these data indicate that lipid overload-induced increased translocation of CD36 to the sarcolemma is an early event prior to the development of insulin resistance and contractile dysfunction (Fig. 4).7 Of note, the pivotal early role of CD36 translocation in this series of events induced by lipid oversupply to the heart was very recently confirmed to occur by another group of investigators.30 Finally, the data further suggest that CD36 is a suitable target to treat myocardial insulin resistance and contractile dysfunction (see below), indicating that the in vitro model described above can be applied to screen for compounds that would impair CD36 function at the sarcolemma.
Fig. 3
Induction of lipotoxic cardiac dysfunction in an in vitro experiment. Isolated rat cardiomyocytes were incubated for 48 hours in a medium with either a low (20 µM) or high (200 µM) concentration of palmitate complexed to bovine serum albumin, in the latter condition in the absence or presence of an anti-CD36 antibody. Cellular triacylglycerol content (left panel) was assessed by thin-layer chromatography, and insulin sensitivity by western blot detection of phosphorylated Akt (middle panel). Peak sarcomere shortening was measured with a video-based imaging system from IonOptix (right panel).
CD36, cluster of differentiation 36.
*Significantly different from control (p<0.05).27

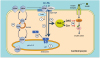
Fig. 4
Mechanism of lipid-induced v-ATPase inhibition and loss of insulin sensitivity in cardiac myocytes upon lipid oversupply. (1) When the supply of LC-FA is low, CD36 is primarily found in endosomes. Furthermore, the v-ATPase V0 subcomplex, which is integral to the endosomal membrane, is assembled with the cytosolic V1 subcomplex, allowing for acidification of the endosomal lumen. In this situation, the available supply of LC-FA is metabolized to meet the immediate energy demands of the myocyte. Elevated extracellular LC-FA supply triggers a series of events: (2a) increased CD36-mediated LC-FA uptake results in elevated intramyocellular LC-FA levels, causing the V1 subcomplex to dissociate; (2b) the V1 subcomplex is shifted toward the cytoplasm; (2c) the disassembly of v-ATPase leads to endosomal alkalinization; and (2d) increased endosomal pH triggers the translocation of CD36 vesicles to the sarcolemma. Upon chronic lipid oversupply, where LC-FA uptake surpasses the metabolic needs of the cell, further processes are set in motion: (3a) the lipid-induced increase in sarcolemmal CD36 feeds forward to further LC-FA uptake and progressive lipid storage (TAG); (3b) excess formation of DAG and Cer culminates into impairment of the insulin signaling cascade and, therefore, loss of insulin sensitivity.35
CD36, cluster of differentiation 36; v-ATPase, vacuolar-type H+-ATPase; LC-FA, long-chain fatty acids; FABPpm, plasma membrane fatty acid-binding protein; FABPc, cytoplasmic fatty acid-binding protein; TAG, triacylglycerol; DAG, diacylglycerol; Cer, ceramides.
2. Tissue-specific approach to manipulate fatty acid utilization
While CD36 has been proposed as a suitable target to rescue the lipid-overloaded heart from contractile dysfunction, targeting its function at the sarcolemma by specific antibodies or inhibitors may not be a preferable systemic approach because such interventions would most likely also impair fatty acid uptake in other organs, especially in adipose tissue, and may affect other known functions of CD36, such as scavenging of oxidized low-density lipoprotein by macrophages.31 Therefore, a preferable approach would be to manipulate the subcellular recycling of CD36, aiming to reduce its presence at the sarcolemma, and if possible, to do so specifically in the heart. Such an approach requires a detailed knowledge of the factors involved in the vesicular trafficking machinery of CD36 recycling between endosomes and the sarcolemma, and of the triggers that regulate the recycling process.
Studies on the intracellular trafficking of various membrane proteins and receptors have revealed that some 50 to 60 distinct proteins are involved in vesicular trafficking pathways, with specific sets of proteins participating in the 3 subprocesses that are commonly identified. Specifically, 1) the fission process involves coat proteins (inducing membrane curvature), Rab proteins (driving unidirectional trafficking via GTP–GDP cycling), vacuolar-type H+-ATPase (v-ATPase; see next section), lipid kinases (locally producing bilayer-destabilizing lipid species), and several adaptor proteins; 2) the translocation process is mediated by motor proteins that transport the vesicles alongside trafficking roads provided by filamentous networks; and 3) the fusion process is mediated by vesicle-associated membrane proteins (VAMPs), Rab proteins, bilayer-destabilizing proteins, and adaptor proteins.3233 Although the CD36-dedicated vesicular trafficking machinery is only beginning to be elucidated, recent reports describing a role played by v-ATPase and specific VAMPs in myocardial CD36 recycling suggest the feasibility of targeting CD36-specific vesicular trafficking to treat lipid overload-induced cardiomyopathy.
3. v-ATPase disassembly and cardiac contractile function
Endosomes, which form the subcellular storage compartment of CD36, are acidic organelles. The low pH (about 5.5) of the lumen of endosomes is maintained through the active import of protons by v-ATPase. Experimental studies with rats fed a high-fat diet have shown that lipid oversupply inhibits the proton-pumping activity of v-ATPase, causing the endosomes to lose their acidification.3435 Importantly, the resulting endosomal alkalinization appeared as early as increased CD36 translocation, and was clearly present before the onset of insulin resistance and the decline in cardiac contractile function. Subsequent studies have revealed that the loss of v-ATPase activity directly caused increased translocation of CD36 from endosomes to the sarcolemma.35
v-ATPase is a multimeric protein complex consisting of 14 subunits. Six of these subunits form the integral membrane subcomplex V0, comprising the proton channel, while 8 subunits make up a peripheral membrane subcomplex V1, which contains the ATP-binding pocket.3637 The functional activity of v-ATPase is assessed by measuring the accumulation of [3H]-labeled chloroquine, a weak base, in isolated cells.34 A main mechanism of regulation of the proton-pumping activity of v-ATPase, originally identified in yeast, is its reversible disassembly into the 2 subcomplexes V0 and V1, whereby the V1 subcomplex may move away from the endosomal membrane to enter the soluble cytoplasm.38 Indeed, the latter mechanism appears to be the cause of v-ATPase inhibition upon lipid oversupply.35 In addition, CD36-mediated lipid uptake is a prerequisite for v-ATPase inhibition, while a consequence of v-ATPase inhibition is the increased translocation of CD36 from endosomes to the sarcolemma.35 The latter observations jointly suggest the occurrence of a feed-forward cycle, in which lipid overexposure increases CD36-mediated lipid uptake, which impairs v-ATPase function, consequently leading to increased CD36 translocation to the sarcolemma and further increased lipid uptake, eventually causing the heart to develop insulin resistance and contractile dysfunction (Fig. 4).35 The corollary is that in cardiomyocytes, endosomal v-ATPase acts like a lipid sensor. However, the molecular mechanism of palmitate-induced v-ATPase disassembly is currently unknown. Searching for interventions that would stabilize the assembly of the v-ATPase subcomplexes V0 and V1 is of interest, as such interventions would form the basis of a novel strategy to interrupt the lipid-induced feed-forward cycle of CD36 translocation and thereby help overcome the detrimental effects of lipid overexposure on cardiac function.
4. VAMP isoforms bring specificity to CD36 trafficking
As with any other protein regulated by translocation, the family of VAMPs has been implicated in the subcellular trafficking of CD36. VAMPs belong to the group of vesicle-associated soluble N-ethylmaleimide-sensitive factor attachment protein receptors, which together with their cognate target-membrane associated SNAREs form so-called SNARE complexes that bring transport vesicles to close enough proximity to the target membrane for the water barrier to be overridden and for fusion of the 2 membranes to occur. A detailed analysis of the various VAMP isoforms present in the heart, seeking to characterize their involvement in insulin-stimulated and/or contraction-induced changes in CD36 translocation, in comparison to that of GLUT4, found 2 VAMPs to be required for both CD36 and GLUT4 translocation (VAMP2 in response to insulin stimulation and VAMP3 in response to electric field stimulation) and 1 VAMP (VAMP4) to be specifically involved in CD36 traffic.39 These differences suggest the possibility of using VAMPs to manipulate specifically CD36-mediated lipid uptake without affecting GLUT4 translocation. The latter point is crucial to allow sufficient glucose uptake and thereby avoid an energy deficit of the heart. VAMP4 would be an especially suitable target for modulating lipid uptake, because targeting VAMP2 and VAMP3 would have similar effects on lipid uptake and glucose uptake. In more detail, inhibiting VAMP4 would block CD36 translocation, and thereby counteract myocellular lipid accumulation and associated lipid-induced cardiac dysfunction. Because VAMP4 is also expressed in skeletal muscle, and has been associated with muscular insulin resistance and type 2 diabetes,40 its inhibition may affect skeletal muscle in a similar manner, limiting muscular lipid accumulation and thereby mitigating whole-body insulin resistance.
CONCLUSION
In this review, we discussed the overwhelming evidence that the transmembrane protein CD36 (SR-B2) plays a pivotal role in the regulation of lipid metabolism, both in the healthy heart and in the development of metabolic cardiac disease. The presence of CD36 in the sarcolemma governs the rate of myocardial lipid uptake, a process that can be seen as the gatekeeper that tunes lipid uptake to the metabolic needs of the cardiomyocyte. In line with this notion, aberrations in the control of CD36 abundance in the sarcolemma—which, in turn, is regulated by the reversible recycling of CD36 between subcellular storage in endosomes and the sarcolemma—appear to constitute the basis of the development of cardiac metabolic diseases, such as diabetic cardiomyopathy. As a result, CD36 can be regarded as a primary therapeutic target to treat metabolic cardiac diseases, not only by lowering lipid uptake in order to combat cardiac lipid overload, but most likely also by increasing lipid uptake in cases of excess utilization of glucose (as seen in cardiac hypertrophy).41 This possibility is underscored by the finding that re-balancing cardiac energy substrate metabolism towards the utilization of an appropriate mix of substrates is a prerequisite for proper cardiac contractile function.42
Insights into the molecular mechanism underlying the subcellular recycling of CD36 between endosomes and the sarcolemma have increased markedly over the last decade, and research in this area has already elucidated the central roles played by v-ATPase and specific VAMP isoforms. Retention of CD36 in the endosomal compartment requires v-ATPase activity, while lipid oversupply decreases this activity. Further research into the mechanism by which lipids (most notably palmitate) induce ATPase disassembly might lead to the design of novel pharmacological agents that reactivate v-ATPase, preferably in a high-lipid environment. Additionally, genetic or pharmacological inhibition of VAMP4 might imprison CD36 within endosomes, thereby restricting cardiac fatty acid uptake. Both strategies may be applied to fight Western lifestyle-induced myocardial insulin resistance and loss of contractile function.
Several aspects of CD36 (patho)physiology still require further study. For instance, at the protein level, the precise mechanism of action of CD36 is not yet clear. The protein undergoes several post-translational modifications, including glycosylation, phosphorylation, and ubiquitination, but whether these are involved in its facilitatory action on lipid uptake remains to be determined.43 In addition, the roles of putative interactions of proteins and/or factors that may influence the functioning of CD36 (e.g., those present in lipid rafts) is still elusive. Interestingly, CD36 is also found in endothelial cells of the heart. Recent studies in rodents suggest that CD36 facilitates transendothelial fatty acid transport through a similar molecular mechanism as outlined above for cardiomyocyte fatty acid uptake.44 Based on these latter findings, it was speculated that in endothelial cells, CD36 may act as a gatekeeper for myocardial fatty acid availability and uptake, thereby indirectly affecting myocardial glucose utilization and insulin action.44 However, in view of the markedly greater abundance of CD36 on the cell surface of endothelial cells compared to cardiomyocytes,45 transendothelial fatty acid transport is not expected to be rate-limiting for overall myocardial fatty acid uptake.
Finally, sarcolemmal substrate transporters are increasingly being recognized as representing the rate-limiting kinetic step of myocardial substrate flux and thus playing a main role in myocardial metabolism and energy provision. As described in the present review, this central role of sarcolemmal substrate transporters for fatty acids and glucose has been established, with major roles played by CD36 and by GLUT1 and GLUT4. The cardiac utilization of alternative substrates (i.e., lactate, ketone bodies and amino acids) is also facilitated by specific membrane substrate transporters, but whether these transporters also regulate substrate uptake is not yet clear.41 In light of increasing evidence for a marked role of these alternative substrates in myocardial homeostatic and pathological processes,4647 such knowledge would be of appreciable interest and may lead to additional targets for metabolic modulation therapy.4849
 XML Download
XML Download