

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
1) When energy needs are high, acyl-CoAs couple to carnitine, which allows them to be imported into mitochondria for beta oxidation and production of heat or adenosine triphosphate.
2) When energy needs are low, acyl-CoAs couple to glycerol, which produces predominant membrane glycerolipids (e.g., phosphatidylcholine, phosphatidylethanolamine, etc.) as well as the triglyceride stores described above.
3) When the energy needs have been met and the storage capacity is full, they can be coupled to a sphingoid backbone derived from the condensation of amino acids and palmitoyl-CoA, producing the sphingolipids.
FUNDAMENTALS OF SPHINGOLIPID BIOSYNTHESIS
Fig. 1
Schematic depicting the key reactions in the ceramide biosynthesis pathway.

• Serine palmitoyltransferase (SPT) condenses palmitoyl-CoA and serine to produce a transient intermediate termed 3-ketosphinganine.3 Occasionally, this enzyme can also use alternative amino acids (i.e., alanine and glycine) to produce deoxysphingolipids.4 The enzyme is multimeric, comprising 2 essential subunits (SPTLC1 and 2) which are requisite for enzyme function, as well as a third (SPTLC3) that can allow myristoyl-CoA to be used in place of palmitoyl-CoA. A family of 3 ORMDL proteins modulate SPT activity; their simultaneous knockdown increases ceramide biosynthesis while their overexpression lowers ceramides.5 The broad spectrum of substrates and components of this enzyme complex suggests that the enzyme complex may serve as a highly regulated gateway for nutrients entering the sphingolipid pool.
• 3-ketosphinganine reductase converts the 3-ketosphinganine produced by SPT into dihydrosphingosine. This reaction proceeds rapidly, such that 3-ketosphinganine levels are exceptionally low. The resultant dihydrosphingosine serves as the sphingoid scaffold that acquires additional, variable fatty acids through the reaction that follows.
• (Dihydro)Ceramide synthases add a variable acyl-CoA to dihydrosphingosine to produce dihydroceramides. The family includes 6 isoforms (CERS1–6) that differ by substrate preference and tissue distribution.6,7 Though CERS2 is the major isoform in the liver, its products, which are very long chain ceramides that include C24 or C24:1 acyl-chains, do not elicit deleterious actions on tissue metabolism.8 The species that most influence anabolic and catabolic processes are the C16 ceramides produced by CERS6, which is upregulated in obesity.9,10 CERS5 also produces C16 ceramides and has been implicated in cardiovascular complications of obesity,11 but does not influence glucose and lipid homeostasis.12 CERS1, which produces C18 ceramides and is the major isoform in skeletal muscle, also influences tissue metabolism and insulin sensitivity.13 CERS3, which also produces really long ceramides, is the major isoform in skin and sperm. As we will discuss below, a recent study suggests that these enzymes have the ability to migrate to the nucleus under conditions of high activity, where they serve as transcriptional repressors of lipase gene expression.14
• Dihydroceramide desaturases insert a conserved double bond into the d4 position of the sphingoid backbone to convert the dihydroceramides into ceramides. This structural feature imparts many of the sphingolipids unique biophysical properties and enables ceramides to serve as regulators of cellular metabolism.2 Two desaturases are present in mammalian tissues, including the ubiquitous isoform DES1 and the tissue-restricted DES2 which is present in the skin and gut. The products of DES2 can contain an additional hydroxyl-group in the D4 position and are termed phytoceramides.
HYPOTHESIS: CERAMIDES ARE EVOLUTIONARILY CONSERVED SIGNALS OF FREE FATTY ACID OVERLOAD
Fig. 2
Schematic depicting the pathways that incorporate fatty acids entering the cell. (A) When detergent-like fatty acid levels get too high, they emulsify cellular membranes. The lysis occurs over 3 stages: fatty acid penetration into the bilayer; their equilibration into both faces of the bilayer; and the transition of the bilayer into a micelle. (B) To prevent lysis, fatty acids entering the cell are quickly coupled to CoA and then incorporated into backbones to produce acylcarnitines, glycerolipids, and sphingolipids. Acylcarnitines are formed as fatty acids translocate into mitochondria. Glycerolipids that are produced make up the bulk of membrane bilayers and create the triglyceride stores that form the majority of the lipid droplet. Sphingolipids are signals of lipid excess that signal to upregulate the other 2 pathways under conditions of lipid excess.

Fig. 3
Schematic depicting the means by which ceramides influence cellular metabolism. Mechanisms described in the text and depicted in greater detail in Fig. 4.

Fig. 4
Schematic depicting the mechanisms of ceramide action.

Ceramides (and SM) facilitate FFA uptake and esterification
Ceramides induce sterol response element binding protein (SREBP) to drive triglyceride formation
Ceramides inhibit glucose and amino acid uptake and utilization
1. Ceramides inhibit insulin and growth-factor stimulated translocation of the anabolic serine/threonine kinase Akt/PKB to the plasma membrane.40 This step precedes the phosphorylation of Akt/PKB phosphorylation on 2 activating residues. The ceramide effector PKCζ is an obligate intermediate in this ceramide action, as it phosphorylates Akt/PKB on a third, inhibitory site in the enzyme's PH-domain.42 The phosphorylation lowers the kinase's affinity for phosphoinositides, thus preventing its PI3-kinase-dependent activation.42 Expression of an Akt/PKB isoform with the S34 site converted to an alanine confers resistance to ceramide in cultured myotubes and vascular smooth muscle.47,48 Ceramides stabilize interactions between Akt/PKB and PKCζ by recruiting the enzymes to membrane rafts or caveolae.48,49
2. Ceramides enhance dephosphorylation of Akt/PKB by activating protein phosphatase 2A.43-46 Inhibition of PP2A, which could be accomplished in cells by either treating with okadaic acid or by overexpressing the SV40 small T antigen, prevents the ceramide inhibition of Akt/PKB in a number of cell types.43-46
Ceramides and ceramide synthases inhibit TAG lipolysis
Ceramides decrease mitochondrial efficiency
The end is nigh: ceramides in apoptosis and fibrosis
Summary of evolutionary basis of these ceramide mechanisms
CERAMIDES DRIVE SELECTIVE INSULIN RESISTANCE, HEPATOSTEATOSIS, AND OTHER CARDIOMETABOLIC DISORDERS
Stage 1: selective insulin resistance
Fig. 5
Schematic depicting the interventions that have been shown to ameliorate cardiometabolic disorders. Pharmaceutical interventions or genetic knockout/knockdown of ceramide-synthesizing genes improves insulin sensitivity, resolves hepatic steatosis, and prevents the development of resultant metabolic disorders including diabetes, cardiomyopathy, coronary artery disease, hypertension, and heart failure.
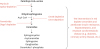
1. Myriocin, the aforementioned inhibitor of SPT, prevents or reverses atherosclerosis,73-78 insulin resistance (i.e., glucose lowering),8,79-83 and hepatic steatosis8,30,79,84-86 in mice, rats, and/or hamsters.
2. Deletion of an essential SPT subunit from adipose tissue of mice resolves glucose intolerance and hepatic steatosis.87
3. Excision of CERS6 from the whole body, liver, or brown adipose tissue of mice improves insulin sensitivity and resolves hepatic steatosis.9,10,12
4. Excision of CERS1 from skeletal muscle of mice resolves insulin resistance.13
5. Excision of DES1 from whole body, adipose tissue, or the liver of mice resolves insulin resistance and hepatic steatosis.2
6. Fenretinide, an inhibitor of DES1, resolves insulin resistance and hepatic steatosis.88
 XML Download
XML Download