

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION OF VASCULAR SMOOTH MUSCLE CELL (VSMC)
1. VSMC plasticity in the vasculature
The fundamental function of fully differentiated VSMCs, which express a wide range of contractile and regulatory proteins, is to maintain arterial blood vessel contractility. VSMCs display considerable plasticity, characterized by reversible switching between contractile and proliferative (i.e., synthetic) phenotypes. In healthy vessels, VSMCs exhibit a contractile phenotype to maintain vascular tone. Contractile VSMCs, which typically show elongated and spindle-shaped morphology, express various smooth muscle-specific contractile proteins including α-smooth muscle actin (α-SMA) and smooth muscle-myosin heavy chain. In contrast, synthetic VSMCs show a stellate shape and have high proliferative and migratory ability, which is essential for the development and repair of damaged vessels.12
In pathological conditions such as vascular injury, arteriosclerosis, and hypertension, fully differentiated VSMCs are able to undergo partial de-differentiation and restart the program of cell growth and proliferation. Owens GK and Schwartz SM have focused on vascular hypertrophy, hyperploidy, and hyperplasia in various forms of experimental hypertension,34 and has suggested that VSMC hypertrophy represents an increase of tissue mass that is an adaptive response to increased functional demands without the loss of any differentiated function. In contrast, VSMC proliferation is related to a transient decrease in the expression of smooth muscle-specific contractile proteins, indicating that synthetic VSMC growth may occur under pathological conditions where functional demands exceed the capacity of VSMCs to respond through cellular hypertrophy.5 This results in hyperplastic remodeling, with less vascular contractility and robustness.67 In atherosclerosis, it has been confirmed by lineage-tracing experiments that VSMCs switch from the contractile phenotype to the proliferative phenotype during atherosclerosis and neointima formation,89101112 and that VSMCs proliferate and further undergo phenotypic switching to phagocyte-like cells, resulting in the accumulation of atherosclerotic plaques and injury-induced neointimal lesions.13141516 Phenotypic switching of VSMCs also reportedly contributes to the development of aortic aneurysms.89101112 A complex regulatory mechanism governs VSMC phenotypic switching through the integration of numerous environmental cues, including cytokines/growth factors, neurohumoral factors, cell–cell contact, cell adhesions, extracellular matrix interactions, injury stimuli, and mechanical forces. In particular, platelet-derived growth factor (PDGF) can dramatically promote multiple aspects of the synthetic VSMC phenotype, while transforming growth factor beta (TGF-β) and its related family members, such as bone morphogenetic protein 4, can increase the expression of VSMC contractile proteins (Fig. 1). Therefore, aberrant hormone release is thought to play a key role in abnormal VSMC plasticity in vascular diseases.
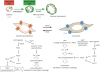
Fig. 1
Characteristics of synthetic and proliferative VSMCs. Contractile VSMCs abundantly express voltage-dependent LTCC, which predominantly mediate Ca2+-dependent contraction through a MLC phosphorylation-dependent pathway (excitation-contraction coupling). In contrast, synthetic VSMCs abundantly expresses receptor-activated TRPC channels, which mediate GPCR-stimulated gene expression through the activation or inactivation of several transcriptional factors (excitation-transcription coupling).
VSMC, vascular smooth muscle cell; LTCC, L-type Ca2+ channels; MLC, myosin light chain; TRPC, canonical transient receptor potential; GPCR, G protein-coupled receptor; α-SMA, α-smooth muscle actin; TGF-β, transforming growth factor beta; PDGF, platelet-derived growth factor; CREB, cAMP response element binding protein; MLCP, myosin light chain phosphatase; MRTF, myocardin-related transcription factor; NFAT, nuclear factor of activated T cells; ROCK, Rho kinase; SRF, serum response factor.
2. Differences in Ca2+ handling in synthetic VSMCs and contractile VSMCs
The transition from contractile to proliferative VSMCs is associated with changes in the expression levels of ion channels, transporters, receptors, and contractile proteins.17 Intracellular Akt-dependent signaling mediates the expression of α-SMA and SM22α proteins, and RhoA mediates actin reorganization, which increases VSMC contractility. In contrast, stimulation of VSMCs with PDGF increases the activity of extracellular signal-regulated kinase (ERK) and cyclin-dependent kinases to promote VSMC proliferation and migration through Krüppel-like family (KLF)-dependent production of matrix metalloproteinases. A major functional difference between contractile VSMCs and synthetic VSMCs is the Ca2+ handling system. Differentiated contractile VSMCs are characterized by rapid transient changes in the intracellular Ca2+ concentration ([Ca2+]i), while the resting cytosolic [Ca2+]i remains low.18 These transient changes in Ca2+ are mainly caused by 2 components of Ca2+ signaling pathways: Ca2+ influx through voltage-dependent L-type Ca2+ channels and dynamic Ca2+ release from intracellular Ca2+ stores. As these Ca2+ dynamics are directed towards VSMC contraction, this pattern is known as excitation-contraction coupling. The synthetic VSMC phenotype is characterized by a long-lasting [Ca2+]i increase (sometimes shown as oscillations) accompanied by a sustained elevation of basal [Ca2+]i.19 During the switch from the contractile to proliferative phenotype, the Ca2+ handling system in VSMCs also transitions from voltage-dependent Ca2+ entry to voltage-independent Ca2+ entry, which is preferentially directed towards gene expression (i.e., excitation-transcription coupling). These changes are associated with altered gene expression, which is dependent on specific transcription factors, such as serum responsive factor (SRF) and SRF-accessory proteins, such as myocardin,1 repressor element 1-silencing transcription factor (also known as neuron restrictive silencer factor)20 and nuclear factor of activated T cells.21 The genes responsible for phenotypic switching in VSMCs include those that code for L-type and T-type Ca2+ channels,2223 voltage-dependent Na+ channels,2425 ClC-3 chloride channels,26 Ca2+-activated K+ channels,2021 inward rectifier K+ channels,2728 Na+-Ca2+ exchangers,29 and canonical transient receptor potential (TRPC) channels.30 Of these various types of channels, TRPC channels have attracted attention as a critical regulator of excitation-transcription coupling induced by various forms of chemical and physical stimulation in the cardiovascular system, and we have recently revealed that a TRPC subtype 6 (TRPC6) negatively regulates TGF-β-induced contractile differentiation in VSMCs (Fig. 2).
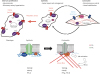
Fig. 2
Physiological and pathophysiological significance of TRPC6 in VSMC phenotype switching. Increased TRPC6 channel activity plays a critical role in determining the VSMC phenotype. Once VSMCs are exposed to metabolic stresses, such as hypoxia, nutrient deficiency and glucose deprivation, TRPC6 channel activity is increased and TRPC6-mediated cation influx suppresses VSMC switching from the synthetic to contractile phenotype upon TGF-β stimulation through PTEN-dependent reduction of Akt activity.
VSMC, vascular smooth muscle cell; TRPC, canonical transient receptor potential; TGF-β, transforming growth factor beta; PTEN, phosphatase and tensin homologue deleted from chromosome 10.
TRANSIENT RECEPTOR POTENTIAL (TRP) CHANNELS IN THE CARDIOVASCULAR SYSTEM
1. TRP channels in VSMCs
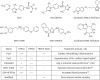
In 1989, Craig Montell first identified the trp gene from a spontaneous mutation of fruit fly Drosophila, which displayed transient elevation of potential in response to light stimuli.31 Since then, extensive studies of this gene have revealed that TRPs make up a superfamily of ion channels that are ubiquitously expressed in mammals, and that 28 members are found in humans. The TRP nomenclature was unified in 2002, and the TRP superfamily is currently subdivided into 6 related protein subfamilies based on genetic and functional similarities: canonical (TRPC1–C7), vanilloid (TRPV1–V6), melastatin (TRPM1–M8), polycystin (TRPP2, P3, P5), mucolipin (TRPML1–L3), and ankyrin (TRPA1). TRP proteins commonly possess 6 transmembrane domains and a preserved sequence of 25 amino acids known as the TRP domain. Despite the high homology among TRPs, it has been revealed that the biophysical features of TRPs are different in terms of activation mechanisms and selectivity. For example, the activity of TRPC family proteins (TRPC1–TRPC7), which are most closely related to the original Drosophila TRP, are polymodally regulated by phospholipase C (PLC)-linked receptor stimulation or other exocytotic mechanisms (Table 1).32 The TRPC3–7 proteins share a high homology (up to 75%) in their amino acid sequence,33 while TRPC1 shows a lower sequence homology than other TRPC members. TRPC1 was initially proposed as a molecular entity of store-operated Ca2+ channels (SOCCs),34353637 which coordinate Ca2+ signaling events in the absence of intracellular Ca2+ stores.38 In VSMCs, however, TRPC1 has been shown to interact with stromal interaction molecule 1, thereby indirectly regulating store-operated Ca2+ entry through the Orai1 channel.3940 TRPC1-dependent store-operated Ca2+ entry has been found to be associated with vasoconstriction in the rat pulmonary artery and caudal artery. TRPC6 also contributes to receptor-stimulated vasoconstriction, but it requires PLC-dependent production of diacylglycerol (DAG), but not inositol-1,4,5-trisphosphate (IP3). The distribution of TRPC subtypes in the cardiovascular system is summarized in Table 1.
Table 1
Molecular characteristics and functional roles of TRPC subtypes in CVS

TRPC, canonical transient receptor potential; CVS, cardiovascular system; PLC, phospholipase C; DAG, diacylglycerol; Ao, aorta; CA, cerebral artery; CoA, coronary artery; PA, pulmonary artery; RA, renal artery; CM, cardiomyocyte; CF, cardiac fibroblast; EC, endothelial cell; RNOS, reactive nitric oxide species; IPAH, idiopathic pulmonary arterial hypertension; 20-HETE, 20-hydroxyeicosatetraenoic acid.
In addition to TRPCs, TRPV (V1, V2, and V4) and TRPM4 subfamily proteins are reportedly present in the cardiovascular system, but the activation mechanisms and electrophysiological properties of TRPVs and TRPMs are quite different from those of receptor-activated TRPC channels.3240 The TRPV subfamily members (V1–V6) contain 3–5 ankyrin repeats within their cytosolic N-terminal region. TRPV1-TRPV4 are all thermosensitive and non-selective cation channels, which show a modest predominance of Ca2+ over Na+ permeation (P
Ca/P
Na = 1–10). TRPV1 is present at high levels in perivascular sensory nerves and participates in the regulation of the tone of small mesenteric resistance arteries stimulated by neuropeptides. TRPV4 channels are also chemosensitive, as they can be activated by cell swelling-induced formation of 5′,6′-epoxyeicosatrienoic acid. As TRPV2 and TRPV4 reportedly participate in cardiac remodeling and endothelium-dependent hyperpolarization induced by mechanical stress, these TRPVs have been proposed to play a role in mechanotransduction in VSMCs. The mechanisms of activation of TRPV5 and TRPV6 are quite different from those of TRPV1–V4, which can be activated by low intracellular [Ca2+]i or hyperpolarization. Proteins in the TRPM subfamily do not contain ankyrin repeats in their N-termini, but have the unique structural feature of a functional enzymatic domain in their C-termini (ADP-ribose pyrophosphatase in TRPM2 and an atypical Ser/Thr kinase in TRPM6/M7). TRPM2 can be activated by an increase in intracellular cyclic ADP-ribose and NAD+, or by hydrogen peroxide. TRPM6/M7 channels are activated by a decrease in Mg2+ concentration. In cerebral arteries, TRPM4 is present, forming a Ca2+-activated non-selective cation channel that regulates myogenic tone regulation (i.e., cerebral blood flow autoregulation).
2. Regulation of TRPC channel activity in VSMCs
As TRP channels are involved in a variety of mechanosensory processes, TRPC channels have also been reported to be sensitive to forms of mechanical stimulation, such as membrane stretching.414243 For example, pharmacological perturbation or gene deletion of TRPC6 has been reported to attenuate excess cardiac contractility stimulated by mechanical stress in Duchenne muscular dystrophy mice.44 Furthermore, it has been demonstrated that the activation of TRPC6 induced by mechanical stretching is mediated by intracellular lipids such as DAG and 20-hydroxyeicosatetraenoic acid (HETE) in A7r5 myocytes (a smooth muscle cell line from rat aorta).4546 As with TRPC6, the TRPC2, TRPC3, and TRPC7 subfamilies have been shown to be directly activated by DAG.4748
The functional roles of TRPC3/C6 have been analyzed primarily with regard to Ca2+ influx and signal transduction in vascular physiology. For example, in the rat portal vein, the TRPC6 channel is activated by an α-adrenergic receptor and evokes membrane depolarization and activation of the voltage-dependent Ca2+ channel, thereby inducing contraction of smooth muscle cells.49 It has been also demonstrated that this TRPC3/C6-induced membrane depolarization can be triggered by vasoactive G protein-coupled receptor (GPCR) ligands, such as angiotensin II and endothelin-1.5051 However, unlike the voltage-dependent Ca2+ channel, TRPC-mediated Ca2+ influx is considered to participate in local Ca2+ signaling, rather than global intracellular Ca2+ mobilization, as TRPC3-mediated local Ca2+ influx is specifically transduced to downstream signaling pathways in B lymphocytes.5253 Furthermore, the TRPC3 protein can interact with various intracellular signaling molecules, including PLC, protein kinase C (PKC), the receptor for activated C-kinase-1, the IP3 receptor (IP3R), and calmodulin,5253545556 suggesting that TRPC3-mediated Ca2+ influx might amplify diverse signaling pathways in the vascular system.
TRPC3/6 channel activity is negatively regulated by Ser/Thr phosphorylation of TRPC3/6 proteins via PKC, protein kinase A (PKA), and protein kinase G (PKG). PKG phosphorylates human TRPC3 at Thr-11 and Ser-263, and human TRPC6 at Thr-70 and Ser-322.57 PKG can be activated by nitric oxide (NO), atrial natriuretic peptide, and inhibitors of cGMP-dependent phosphodiesterases (PDEs). The PKG-dependent suppression of TRPC6 channel activity by NO is physiologically important in endothelium-dependent vasodilation.58 Both PKA and PKG can recognize the same substrate sequence (-R-R/K-X-S/T-), and PKA-dependent phosphorylation of rodent TRPC6 at Thr-69 has been shown to participate in endothelium-independent vasodilation.51 Increased PKG activity reportedly suppresses Ca2+/calcineurin-dependent cardiac hypertrophy induced by receptor stimulation and pressure overload, and blockade of PKG-dependent phosphorylation by TRPC6 mutagenesis (i.e., substitution of Thr-69 to Ala) reversed the PKG-dependent anti-hypertrophic action.59 In contrast, reduction of cGMP/PKG signaling by guanylate cyclase-A gene deletion caused spontaneous cardiac hypertrophy by promoting TRPC3/6 channel activity.60
3. Vascular tone regulation by TRPC6
The physiological importance of TRPC6 in vascular mechanosensation has been revealed in TRPC6-deficient mice.61 An increase of blood pressure inside the small arteries unexpectedly results in vasoconstriction. This phenomenon, which is known as the Bayliss effect, involves the stretch-activated non-selective cation channels in VSMCs.6263 TRPC6 deficiency decreases VSMC contraction and depolarization induced by pressure in arteries; for instance, the basal mean arterial pressure in TRPC6-deficient mice is approximately 7 mmHg higher than that in wild-type mice.61 It has been recently reported that the stretch-induced channel activity originates from a fascinating interplay between the TRPC6 channel and Gq/11 protein-coupled GPCRs, including angiotensin II type 1 receptor (AT1R).64 In fact, the myogenic tone induced by an increase in intravascular pressure was attenuated in AT1R-deficient mice.65 Since TRPC6 also functionally couples with Gq/11PCRs that are responsive to membrane stretching,4346 TRPC6 is widely accepted to be a mechano-activated cation channel. TRPC6 deficiency causes acute arterial hypoxemia in the mouse pulmonary artery,66 a phenomenon known as the Euler-Liljestrand reflex in the small pulmonary artery.67 This process includes accumulation of intracellular DAG and reactive oxygen species (ROS) via NADPH oxidase 2 (Nox2) activation in endothelial cells, leading to an increase in endothelial permeability and edema formation.45 In patients with idiopathic pulmonary arterial hypertension, certain single-nucleotide polymorphisms in the TRPC6 gene promoter have been shown to be associated with enhanced expression of TRPC6 mRNA and protein.6869 Enhanced expression of TRPC6 caused pulmonary arterial smooth muscle cells to switch from a contractile to synthetic phenotype via an increase in [Ca2+]i by upregulating store-operated Ca2+ entry.70
4. Role of TRPC channels in VSMC plasticity
Contractile VSMCs abundantly express the large-conductance Ca2+-activated K+ channel, a voltage-dependent channel that is activated by membrane depolarization.71 The function of this channel is to provide negative feedback against membrane depolarization, limiting voltage-dependent Ca2+ influx through L-type Ca2+ channels, a high-affinity site of action of anti-hypertensive Ca2+ channel blockers.72 The switch of VSMCs to the proliferative phenotype is associated with loss or suppression of both voltage-dependent channels.73 However, we should not exclude the possibility that these channels could contribute to very early events in responses to injury74 or return once the modulated cells have ceased their activity and begun a more quiescent existence. That is, timing is a critical factor, meaning that it would be wrong to oversimplify the situation by assuming the cells either have a pure contractile phenotype or are constantly in a proliferative phenotype.75
In contrast, proliferative VSMCs lose voltage-dependent channels, while enhancing native store-operated or receptor–activated Ca2+ entry that fails to couple with contraction.76 These channels are necessary for receptor-stimulated cell proliferation or migration, and several TRP channel proteins have been identified as critical components of receptor-activated cation channels in VSMCs.75 TRPC1 is reportedly upregulated in response to vascular injury, and inhibition of TRPC1 attenuates VSMC proliferation7778 and hyperplasia induced by angiotensin II.79 TRPC1 also exerts a robust functionality by forming heteromultimers with TRPC580 and its related isoform TRPC4.81 Since TRPC5 and TRPC4 are predominantly expressed in endothelial cells, but not in VSMCs, these heteromultimers may play a pivotal role in endothelial cells.
Indeed, the most important difference between the 2 VSMC phenotypes is that contractile VSMCs express more abundant contractile proteins, such as α-SMA and SM22α than synthetic VSMCs.1 PDGF is a major cytokine that contributes to mural cell recruitment to capillaries, while TGF-β is a major cytokine involved in the contractile differentiation of VSMCs.82 We have recently reported a signaling mechanism through which TRPC6 channel activity negatively regulates contractile differentiation in VSMCs induced by TGF-β stimulation.83 Using TRPC6-deficient VSMCs, we found that TGF-β-induced contractile differentiation in VSMCs was significantly enhanced compared to wild-type VSMCs, while PDGF-induced proliferation and migration of VSMCs were unaffected. As the background intracellular Ca2+ entry was not significantly different between wild-type VSMCs and TRPC6-deficient VSMCs, we focused on the plasma membrane potential, because more quiescent and fully differentiated cells reportedly exhibit relatively polarized membrane potentials, ranging from −50 to −90 mV, whereas more plastic stem cells exhibit less polarized membrane potentials, averaging from −10 to −40 mV.84 Indeed, contractile TRPC6−/− cells showed more hyperpolarized membrane potentials than contractile TRPC6+/+ cells. Simultaneously, TGFβ1 stimulation increased SM22α expression to a higher level in TRPC6−/− cells than in TRPC6+/+ cells. By using C3H10T1/2 mesenchymal stem cells as a well-defined VSMC differentiation model, we found that knockdown of TRPC6 significantly increased TGF-β1-induced activation of Akt, a major mediator of VSMC differentiation accompanying the upregulation of contractile proteins.85 TRPC6 has been reported to interact with phosphatase and tensin homologue deleted from chromosome 10 (PTEN), a negative regulator of Akt, by inhibiting the production of phosphatidylinositol (3,4,5)-trisphosphate, in endothelial cells and VSMCs, and this interaction is important for cell surface expression of TRPC6.8687 We also confirmed that TRPC6 interacts with PTEN, through a process in which TRPC6-mediated cation influx negatively regulates Akt activity via membrane depolarization. Changes in membrane potential influence the distribution of phosphatidylserine (PS) in the plasma membrane.88 PTEN possesses a C2 domain that is critical for its translocation to the plasma membrane and enzymatic activation. The C2 domain binds to anionic phospholipids such as PS and phosphatidylinositol in a global Ca2+-dependent manner. Thus, TRPC6-mediated cation influx may cause membrane depolarization, followed by voltage-dependent Ca2+ influx through L-type Ca2+ channels, thereby downregulating the differentiating activity of VSMCs through PTEN-mediated Akt inactivation (Fig. 2). However, the TRPC6 channel does not conduct a large amount of current. Therefore, it is not likely that TRPC6-mediated cation influx rapidly depolarizes the membrane potential of VSMCs. As TRPC-dependent currents are long-lasting, longer-time imaging of membrane potentials in VSMCs will be necessary to elucidate its underlying mechanism.
THERAPEUTIC POTENTIAL OF TRPC CHANNELS IN CARDIOVASCULAR DISEASES
Therapeutic applications of TRPC channels have been well studied using cardiac systems. Structural and morphological changes (remodeling) of the heart are a clinical outcome of heart failure, and many studies have shown that TRPC3 and TRPC6 are involved in the development of cardiac remodeling.4459608990 Cardiomyocyte-specific overexpression of TRPC3 or TRPC6 is hypersensitive to the hemodynamic load and promotes cardiac hypertrophy in mice.9192 Since knockdown of either TRPC3 or TRPC6 completely suppressed angiotensin II-induced hypertrophic growth in rat cardiomyocytes89 and the inhibition of TRPC3 or TRPC6 channel activity significantly attenuated cardiac hypertrophy in mice in vivo,44 many researchers—including our group—have concluded that TRPC3 and TRPC6 heteromultimer channels clearly participate in the development of cardiac hypertrophy.93 Based on these observations, several small molecules that can inhibit TRPC3/C6 channel activity were revealed to suppress pathological cardiac remodeling in mice (Fig. 3). In particular, our in vivo studies using TRPC-deficient mice have revealed that TRPC3, but not TRPC6, predominantly participates in pressure overload-induced cardiac remodeling—especially in interstitial fibrosis, but not in myocardial hypertrophy.9495 We have also found that TRPC3 has little impact on myocardial global [Ca2+]i under mechanically stretched conditions, but significantly suppresses mechanical stretching-induced ROS production. The TRPC3 protein interacts with Nox2 on the plasma membrane, which enables Nox2 to escape from ER-associated degradation, leading to excess ROS production in both cardiac myocytes94 and fibroblasts95 and resulting in the induction of interstitial fibrosis through the respective signaling pathways. Although it has been shown that the TRPC6 protein is upregulated in pathological rodent hearts,92 TRPC6 has been found to counteract the TRPC3-Nox2 protein complex and to abrogate Nox2-dependent ROS signaling in cardiomyocytes.96 This evidence suggests that TRPC6 upregulation is an adaptive response against environmental stress in order to avoid inducing excess ROS production (i.e., oxidative stress) in the heart. Indeed, among several TRPC3 inhibitors, only pyrazole-3 can suppress the TRPC3-Nox2 axis-dependent cardiac stiffness and atrophy caused by anthracycline-derivative anti-cancer drug treatment.90 We recently screened a potent inhibitor of the TRPC3-Nox2 complex using a library of already approved drugs and found that ibudilast, a PDE4 inhibitor approved for the treatment of asthma and dizziness secondary to chronic cerebral circulation impairment associated with the sequelae of cerebral infarction, significantly suppressed doxorubicin-induced atrophic shrinkage of cardiomyocytes and skeletal muscles, as well as macrophage cell death.9798 However, our preliminary experiments indicate that TRPC3 selective inhibitors have little impact on VSMC plasticity or vascular diseases, including peripheral arterial disease. This may be because TRPC6 is highly expressed in VSMCs and predominantly regulates VSMC plasticity, rather than the TRPC3 channel or the TRPC3-Nox2 protein complex. Indeed, several small molecules that can inhibit TRPC6 channel activity are able to reduce blood pressure and pulmonary artery hypertension.5199100 These observations suggest that inhibition of TRPC6 channel activity is a promising strategy for the treatment of aberrant VSMC plasticity in vascular diseases.
FUTURE PERSPECTIVES
Accumulating evidence has suggested that the expression of TRPC1 and TRPC6 in VSMCs can be exploited to control systemic arterial blood pressure and local blood flow, and the manipulation of TRPC1/TRPC6 channel activity offers a new therapeutic strategy for the treatment of lung ischemia-reperfusion injury.101102 We recently reported that TRPC6 participates in the negative regulation of VSMC contractile differentiation under pathological (ischemic) conditions. Although upregulation of the TRPC6 protein is required for the efficient physiological proliferation of VSMCs, it is still obscure why the TRPC6 protein is continuously upregulated in pathological VSMCs, and whether TRPC6 also participates in aberrant VSMC proliferation in human vascular diseases. Further spatio-temporal analyses of TRPC6 protein function are necessary to establish the pathological significance of TRPC6 in VSMC plasticity. In addition, as TRPC6 is also expressed in endothelial cells and mediates endothelial permeability in capillary microvessels in response to histamine stimulation,103 it is also necessary to investigate the roles of TRPC6 in non-VSMCs, such as endothelial cells and macrophages, to achieve a comprehensive understanding of the clinical significance of TRPC6 channels in the vasculature.
Furthermore, more attention should be paid to the diverse patterns of cation permeability shown by TRPC channels (Table 1). Unlike TRPC3, divalent metal cations such as iron (Fe2+) and zinc (Zn2+) can permeate through TRPC6, even though TRPC3 and TRPC6 are up to 75% identical.104105 Indeed, TRPC6−/− mice presented an elevated Zn2+ level in the placenta and reduced litter sizes.106 Although both Zn2+ and Fe2+ must be essential metal cations for the maintenance of cellular homeostasis, the causal relationship between metal cation dynamics and vascular plasticity is obscure. Future studies focusing on TRPC6-mediated metal ion influx and identification of its relationship with VSMC plasticity will help us to obtain a comprehensive understanding of how TRPC6 can serve as a specific therapeutic target for pathological VSMC plasticity.
 XML Download
XML Download