

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Our body is exposed to various stimuli from the external environment. Due to direct exposure to the environment, and with a large surface area, the skin and gastrointestinal tract encounter various pathogens and function as the primary barrier to protect the host.1 If a pathogen penetrates this barrier, the special defense system of its host, i.e., the immune system, is activated.2 As the defense employed to protect the host from external invaders, the immune system is often compared to an army. The immune system comprises humoral immunity and cell-mediated immunity.3 B cell-based humoral immune cells produce a variety of antibodies to neutralize extracellular pathogens. Cytotoxic lymphocytes, cytotoxic T cells, and natural killer cells contribute to cell-mediated immunity. Cytotoxic lymphocytes distinguish between normal cells and abnormal cells, such as pathogen-infected host cells, and trigger the death signal in those infected cells.3 Besides fighting infection, immune cells induce the death of ‘strange’ host cells and maintain host homeostasis. Various ligands of normal host cells correspond to receptors on cytotoxic cells; thus, ligand-receptor binding suppresses the activation of the cytotoxic cells.4 Cytotoxic lymphocytes recognize selfness through their own receptor activity. In the case of different surface antigens being presented, cytotoxic cells are activated, this being the main contribution to the cytotoxic clearance of the infected cell or the non-self cell (i.e., the transplanted organ). 34 Because cell mediated immune function can directly induce cell death, the cytotoxic clearance mechanism is tightly regulated by multiple steps.5 Paradoxically, this self-sorting immune check system allows malignant cancer cells to evade the cell-mediated immune reaction.6 Recently, engineered T cells that can specifically target cancer antigens, chimeric antigen receptor T cells (CAR T cell), have emerged as novel immunotherapeutic modality.
IMMUNE ESCAPE OF CANCER
An accumulation of somatic mutations is a common feature of malignant cancer. Although most somatic mutations are phenotypically silent, random somatic mutations allow functional benefits, which confer a survival advantage.7 For example, the K-Ras mutation allows continuous binding to guanosine-5′-triphosphate without distinct triggers and induces an increase in cell-cycle turnover. This is the most frequently observed mutation in the development of colorectal cancer from preneoplastic lesions.8 Cytotoxic lymphocytes also recognize ‘strange’ cells that develop as a result of somatic mutations; therefore, the tumor cell can be cleared under the surveillance of the immune system.9 However, an accumulation of somatic mutations can dissimulate their identity through the loss of antigenicity.10 In this way, cytotoxic leukocytes can fail to recognize an unhealthy cell because it shows normal surface antigens. Besides the passive mechanism of loss of antigenicity, cancer cells can actively incapacitate the immune system. Cancer cells generate immune modulating factors on their cell membrane to evade cytotoxic T cell recognition.10 PD-L1 binds to the PD-1 receptor11 and the B7 factor12 specifically binds to CTLA-4 on a cytotoxic T cell. Ligand binding of PD-1 or CTLA-4 inactivates cytotoxic T cells, which results in immune escape.10 Furthermore, cancer cells indirectly suppress the cell-mediated immune system. Cancer cells actively secrete immunosuppressive molecules such as those from the galectin family,13 the CXCL family,14 or inhibitory interleukins (e.g., IL-10).15 These are released from cancer cells and activate various immune regulatory cells such as myeloid-derived suppressor cells, regulatory T cells, tumor-associated macrophages, or tumor-associated neutrophils. These cells neutralize the cytotoxic immune response and thereby permit cancer cell survival and expansion. In short, cancer cells actively generate immune escape tools to survive in the zones of massive inflammation.6
CHIMERIC ANTIGEN RECEPTOR T CELL
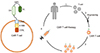
Although abundant immune cells are accumulated in the cancerous region, the immune system deteriorates.6 Cancer cells interfere with the activation of cytotoxic T cells. Cytotoxic T cell activation requires multiple fastidious steps to avoid abnormal activation, this tight regulation granting an adverse immune evasion opportunity to the cancer cell.16 Even if only one activation molecule is missing from the signaling pathway, the T cell will not be activated. Cancer cells can easily incapacitate the host immune system simply through inhibition of one ligand of the T cell.16 Hence, many researchers have trialed methods to recover the cell-mediated immune response. The goal of these engineered cells is to specifically target cancer cells and overcome the immunosuppressive microenvironment created by them.171819 To specifically target cells of malignant cancer, antibodies that target cancer cells are utilized. For the simplification of therapy, diverse but essential factors for T cell activation are combined into a single receptor. Cancer-specific recognition parts are fused to form the external part of the receptor. This T cell, which carries a chimeric antigen receptor on its cell membrane, is known as the chimeric antigen receptor (CAR) T cell.19 Fig. 1 demonstrates CAR T cell generation and elucidates its structure.
CAR T cell therapy shares clinical features with monoclonal antibody therapy.20 Extremely specific ligands for antigen recognition are mandatory for CAR T cell generation, which is mostly adopted from the antigen binding motif of monoclonal antibodies. Monoclonal antibody therapy is an emerging strategy to target not only malignant cancer but also chronic refractory disease.21 The primary characteristic of a monoclonal antibody is that it is a macromolecule and its therapeutic potential largely depends on the flow of the bloodstream, which means a monoclonal antibody is hardly ever delivered to hypoxic areas.22 Furthermore, the monoclonal antibody itself could possibly trigger an immune response in the host, resulting in therapeutic failure of the neutralizing antibody.23 In contrast, as a CAR T cell is a “cell” active mobility and target recognition is easily achieved.24 The cells used in CAR T cell therapy are originally obtained from the host, then modified to acquire chimeric antigen receptors. Therefore, secondary immune activation is rarely induced.25
To transduce the chimeric antigen-presenting gene, a genomic incorporation virus, retrovirus, or lentivirus is commonly used.26 Genetic engineering, by use of a retrovirus or lentivirus, has several benefits. Retroviruses and lentiviruses easily infect mammalian cells and are incorporated into the host genome. Furthermore, both viruses minimally affect host cells after infection and consequent genomic incorporation. Their most powerful advantage is that exogenous genetic material incorporated by viral infection is also amplified when host cells divide, ensuring we can provide the appropriate number of cells, despite an initially limited number of infected cells.27 Although retroviral vectors are known to be a safe modality in experimental conditions, available evidence remains unclear. Moreover, we are not able to control the incorporation locus of the viruses, which implies that certain essential genomic loci in host cells may be disrupted by exogenous genes.28 Recently, the CRISPR/Cas9 technique-based integration of the CAR gene has emerged and the greatest advantage of the CRISPR/Cas9 system is that the target locus in the host genome can be strictly regulated. For safety, genomic incorporation of AAVS1, CCR5, and ROSA26 has been tested.29 Nevertheless, CAR T cell therapy is a new, emerging technology, and its long-term efficacy and adverse reactions should be further investigated.30
CLINICAL TRIALS AND ADVERSE REACTIONS
A CAR T cell needs to bind to a specific surface target. Hence, CAR T cell treatment can work well on a malignant cancers that present unique antigens. The earliest CAR T cell trial targeted CD19 positive leukemias, such as CD19+ acute lymphoblastic leukemia or CD19+ diffuse large B cell lymphoma.31,32 More than 300 clinical trials are ongoing to evaluate the therapeutic potential of CAR T cells in various cancers, especially for cancers with poor prognoses. (https://clinicaltrials.gov/ct2/home).
Only two CAR T cell therapies have so far been approved. Both are targeted towards CD19+ B cell malignancy. Tisagenlecleucel (trade name Kymriah®) received approval from the U.S. Food and Drug Administration (FDA) in August 2017.33 Kymriah is approved for CD19 positive acute lymphoblastic leukemia and relapsed diffuse large B cell lymphoma. As per the recommendations, it is indicated in patients up to 25 years of age with refractory or second/later relapse large B cell precursor acute lymphoblastic leukemia.33
Axicabtagene ciloleucel (trade name Yescarta®) is another CAR T cell therapy approved by the FDA in October 2017.34 Yescarta is indicated as a second-line treatment for diffuse large B cell lymphoma, primary mediastinal B cell lymphoma, and transformed follicular lymphoma. According to the clinical trial ZUMA-1, Yescarta can be prescribed for the treatment of non-Hodgkin's lymphoma.
CAR T cell treatment has been known to induce adverse reactions; common adverse reactions of CAR T cell therapy include cytokine release syndrome and neurological toxicity.25 CAR T cells elicit an immune response. Although a CAR T cell is generated from a host cell, it can be recognized as a foreign molecule, stimulate an immune response, and thereby induce an anaphylaxis-like immune response.35 Hence, CAR T cell infusion provokes massive inflammation and results in a surge of cytokine production; the most common adverse reaction being cytokine release syndrome. Fortunately, a monoclonal antibody, tocilizumab (trade name Actemra®) is approved to control cytokine release syndrome after CAR T cell infusion.17
Neurological toxicity is also commonly observed, but its specific mechanism remains unclear.36 It is considered to occur independent of the cytokine release syndrome. Mild symptoms such as delirium, expressive aphasia, and obtundation, as well as severe adverse effects such as death from cerebral edema, have been reported.37 Although most of the neurological adverse reactions are temporary and reversible, CAR T cell infusion should be closely monitored for adverse reactions.
CAR T CELLS IN CANCER TREATMENT
To date, therapeutic outcomes with CAR T cells have mostly been acquired in non-Hodgkin's lymphoma, because approved CAR T cell treatments target CD19+ B cells. CAR T cell therapy achieved about 70% primary remission and more than 50% of patients acquired complete remission.38 The most promising potential of CAR T cell therapy is that it expands therapeutic options for refractory patients undergoing conventional treatment. Diffuse large B cell lymphoma (DLBCL) is the most common subtype of B cell-related lymphoma and rituximab, an anti-CD20 monoclonal antibody-based chemotherapy, is a well-established strategy. Approximately 50% of DLBCL patients experience complete remission with conventional chemotherapy, but unfortunately, approximately 30% of patients with complete remission by chemotherapy also experience relapse. Furthermore, 20% of DLBCL cases experience primary refractory after chemotherapy.39 CAR T cell achieved 50% complete remission with minimal side effects among patients with primary refractory or relapsed DLBCL by rituximab-based chemotherapy.40
THERAPEUTIC POTENTIALS BEYOND CANCER AND FUTURE PERSPECTIVES
CAR T cell therapy was originally designed to target surface antigens of cancer cell membranes and thereby erase cancer cells; this gives rise to a fundamental question of whether a CAR T cell had the capacity to remove an undesirable non-cancer cell. Recently, Aghajanian et al.41 reported the therapeutic potential of CAR T cells against cells other than cancer cells. First, they generated mice that artificially overexpressed ovalbumin peptide (OVA) in fibroblasts under tamoxifen treatment. Xenogeneic ovalbumin peptide production was controlled under a postin-promoter that was specifically upregulated in activated fibroblasts and not quiescent fibroblasts. The mice were subjected to a continuous infusion of angiotensin II and phenylephrine for 4 weeks, which robustly aggravated myocardial damage and caused subsequent cardiac fibrosis. Next, the mice were infused with OVA peptide cognate CD8+ T cells (OT-I CD8+ cells) 1 week after treatment with fibrogenic agonists. Allogeneic transplantation of OT-1 CD8+ cells dramatically ablated the interstitial fibrosis generated by angiotensin II and phenylephrine without significant adverse effects. Based on heat map analysis, fibroblast activation protein (FAP) was determined as the endogenous target for treating fibrosis. Finally, the capability of CAR T cells to target FAP was measured. As in the trial of OT-1 CD8+ cells, FAP CAR T cells were adopted 1 week after cardiac damage and boost transplantation was performed a week later. Massive cardiac fibrosis, measured by Sirius Red staining, was dramatically reversed in the FAP CAR T cell-treated mice. Cardiac dysfunction correlated with interstitial fibrosis had also improved in the FAP CAR T cell mice with evidence of minimal inflammation in various organs.41
CAR T cell approaches can be used to neutralize autoimmune diseases. In autoimmune conditions, antibodies target host cells.42 Memory B cells carrying autoantibodies attack the host. With respect to cell therapy, abnormally activated B cells are the main target for clearance. CAR T cells were originally designed to express antibody-like domains to claim a “cancer antigen”. However, for the CAR T cells to incapacitate an autoimmune disease, they should present antigens recognized by autoimmune B cells. Ellebrecht et al.43 reported the therapeutic potential of CAR T cells in autoimmune dermatitis, Pemphigus vulgaris (PV). Whole B cell ablation by conventional CAR T cells, which targeted CD19+ B cells, resulted not only in an immunocompromised period but also showed notable rates of recurrence. Hence, they designed CAR T cells to specifically target autoimmune antibody-producing B cells. Experimentally, T cells were modified to express the Dsg3 ‘antigen,’ the main target of keratinocytes in PV. The cells were renamed as chimeric autoantibody receptor (CAAR) T cells. CAAR T cell transplantation into the mouse model of PV induces disease remission with a lower rate of recurrence and a minimal risk of an immunocompromised state.43
The most promising feature of CAR T or CAAR T cell treatment is its expandability (Fig. 2). If the antigen binding motif changes, a different cell is targeted therefore, monoclonal antibody therapy can be applied to CAR T treatment. Monoclonal antibody therapies are also designed to target specific human antigens such as cancer antigens (rituximab),44 surface receptors that require fast growth (cetuximab)45 or signal activation (omalizumab).46 They also bind to extremely specific antigens of host cells, which indicates that monoclonal antibodies and their human antigens are already proven therapeutic targets in those diseases. As briefly mentioned above, CAR T cells require engineered surface chimeric antigens that can bind to the cancer antigen. CAR T cell treatment can be applied to human disease by designing CAR T cells to express different single chain fragments of the antibody.47 For example, FAP is not limited to cardiac fibrosis, but is generally expressed in activated fibroblasts.48 In simple terms, FAP CAR T cells could be applied to fibrosis-associated diseases that cannot be controlled by conventional therapeutic approaches. Perinatal infection of hepatitis B virus mainly contributes to chronic hepatitis and hepatic fibrosis, which eventually accelerates hepatic cellular carcinoma.49 In fact, hepatic fibrosis, also known as liver cirrhosis, develops as a result of a normal immune reaction. Virus-infected hepatocytes are subjected to a cell-mediated immune reaction. Unfortunately, the immune reactive death of the infected hepatocytes is rarely effective in clearing the virus, because the released hepatitis virus infects the neighboring hepatocytes. A healthy immune system paradoxically activates a vicious chronic inflammation reaction, which triggers fibrotic changes. Physicians are still only focused on handling the hepatitis virus. However, the complications of liver cirrhosis include portal vein hypertension, peripheral edema, varicose bleeding, and jaundice.50 CAR T cells could be considered a future modality to improve cirrhosis in these types of cases.
Idiopathic pulmonary fibrosis (IPF) is a representative life-threatening fibrosis-associated disease.51 Although the specific underlying mechanism remains unclear, viral infection or exposure to hazardous gases such as air pollutants or cigarette smoke, are indicated as possible causes. Chronic progressive inflammation finally destroys the pulmonary parenchyma, which is essential for gas exchange; subsequently, the damaged cells are filled with extracellular matrix in the process of fibrosis.51 The current therapeutic strategy for IPF also focuses on the modulation of inflammation, but it does not reverse fibrosis issues.52 With consideration of the outcomes in cardiac fibrosis, CAR T cells could be considered to ameliorate the fibrosis issue in IPF.
The indications of CAAR T cells can also be expanded to various autoimmune diseases,43 such as myasthenia gravis, Hashimoto's thyroiditis, systemic lupus erythematosus, rheumatoid arthritis, ankylosing spondylitis, multiple sclerosis, psoriasis, and type 1 diabetes mellitus. Most autoimmune diseases are controlled using immune modulators such as nonsteroidal anti-inflammatory drugs, glucocorticoids, or even mild cytotoxic chemicals to nonspecifically erase immune cells. Based on the CAAR T cell potential for PV, many autoimmune diseases could be included in the future indications of CAAR T cell treatment. Unlike CAR T cells, CAAR T cells express an ‘antigen’ molecule, which means CAAR T cells can activate the humoral immune reaction. Large amounts of antigen, which are attached to a CAAR T cell surface to recognize pre-existing memory B cells, might result in an immense immune reaction. De novo antibody formation can also be considered. Fortunately, no adverse response of the CAAR T cell therapy has been reported to date.43 Physicians should closely monitor for potential adverse effects when employing CAAR T cell treatment in the future.
CONCLUSION AND LIMITATIONS
A CAR T or CAAR T cell is a T cell obtained from the patient and engineered to serve as personalized medicine. CAR T cells cannot be a ready-to-use regimen. In terms of the commercial aspect, this kind of tailored therapy could pose a massive financial burden on the patient.53 A CAR T cell also requires an extremely limited, but specific object to target. Hence, CAR T cell therapy shares some clinical features of monoclonal antibody therapy.54 However, monoclonal therapy can be commercialized more easily. Thus, the cost-effectiveness aspect of CAR T cell treatment is a critical hurdle in the development of the clinical indications of CAR T cell treatment.53
For CAR T cell therapy, a specific target is mandatory. However, in many cancers, the cells do not present a unique antigen.55 Genetic variety of the human genome results in the possibility of different antigens being similar, implying that CAR T cell therapy for cancer antigen requires fine modulation depending on the patient's polymorphisms. This modulation might worsen clinical applications in the future.

 XML Download
XML Download