

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Soluble epoxide hydrolase (sEH, EC 3.3.2.10) is the major enzyme responsible for the hydrolysis of epoxy fatty acids (EpFAs) to their corresponding vicinal diols in humans and other mammals.1 These EpFAs include the epoxides of linoleic, arachidonic, eicosapentaenoic, and docosahexaenoic acid that are produced primarily by cytochrome P450s. These natural molecules are pleiotropic endogenous mediators with key functions in inflammation,1 pain,2 and blood pressure regulation.3 Increasing the levels of endogenous EpFAs by inhibiting sEH has been shown to block and resolve inflammation,4 reduce pain,2 lower blood pressure, and prevent cardiovascular diseases.5 To overcome these problems, finding new inhibitors from natural plants has been investigated. A few sEH inhibitors from natural products have been identified. The results showed that natural compounds found to inhibit sEH were diverse including biflavonoids,6 selaginellin,7 stilbenes,8 anthraquinone derivatives,9 carbazole-type alkaloids (isomahanine, bisisomahanine),10 alkylphloroglucinol derivatives, and triterpenoids.11 These results encourage us to continue our studies in discovery of sEH inhibitors from natural sources.
From our screening results, we found that the ethanol extract of Passiflora edulis Sims had appreciable inhibitory activity. Passiflora edulis Sims (Passifloraceae), a popular tropical fruit throughout the world, is usually used for juice production,12 and widely cultivated in South America, Africa, and Asia. In Vietnam, P. edulis is popularly cultivated in Tay Nguyen, Nghe An and Son La with areas of over ten thousand hectares. P. edulis was found to possess biological activities including anti-inflammatory,13 antihypertensive,14 anti-oxidant,15 anti-tumor,16 anti-anxiety,17 antifungal,18 and found to inhibit melanogenesis and promote collagen synthesis.19 Previous studies on chemical constituents of P. edulis showed the presence of triterpenoids,20 flavonoids,21 alkaloids,22 carotenoids,23 stilbenoids,1924 oil, and tocopherols.25 In spite of the number of studies that have been performed,192425 there has been no investigation of chemical constituents and sEH inhibitory activity of P. edulis seeds cultivated in Vietnam. Therefore, this paper described the isolation and structural elucidation of these compounds as well as the evaluation of their inhibitory activity on sEH.
Experimental
General experimental procedures
1H-NMR (500 MHz) and 13C-NMR (125 MHz) were measured on a Bruker Avance 500 MHz spectrometer. ESI-MS was obtained from a Varian FT-MS spectrometer and MicroQ-TOF III (Bruker Daltonics, Ettlingen Germany). Optical rotations were measured on P-2000 polarimeter (JASCO, Tokyo, Japan). Column chromatography was carried out on silica gel (Si 60 F254, 40-63 mesh, Merck, St. Louis, MO, USA). All solvents were redistilled before use. Precoated TLC plates (Si 60 F254) were used for analytical purposes. Compounds were visualized under UV radiation (254, 365 nm) and by spraying plates with 10% H2SO4 followed by heating with a heat gun.
Plant materials
The seeds of Passiflora edulis Sims were provided by Nafoods Group JSC (Nghe An Province, Vietnam) in 2016 and identified by botanist Dr. Nguyen Quoc Binh, Vietnam National Museum of Nature, VAST, Hanoi, Vietnam. A voucher specimen (C-573) was deposited in the Herbarium of the Institute of Natural Products Chemistry, VAST, Vietnam.
Extraction and isolation
The dried powdered seeds (1.0 kg) of P. edulis were extracted three times with n-hexane (3 × 4.0 L) at room temperature for 3 days, filtered, and then concentrated under decreased pressure to give n-hexane extract (200 g) and residue. The dried residue (700 g) was then extracted three times with ethanol (3 × 3.0 L) by sonication for 6 hours. The ethanol extract (60 g) was suspended in hot-water (0.3 L) and partitioned with dichloromethane (CH2Cl2, 3 × 3.0 L) and ethyl acetate (EtOAc, 3 × 3.0 L) successively. The resulting fraction was concentrated under decreased pressure to give CH2Cl2 (5.2 g) and EtOAc (20 g) fractions, respectively. By the guided-fractionation activity, the EtOAc soluble fraction was chromatographed on a silica gel column chromatography (CC) eluting with a gradient of CHCl3–MeOH (20:1 to 0:1) to afford eight fractions (Fr. E1 to Fr. E8). Fraction E3 (820 mg) was subjected on a silica gel CC eluting with a gradient of n-hexane–acetone (4:1 to 0:1) to afford compounds 1 (6.2 mg) and 2 (200 mg). Fraction E4 (3.5 g) was also subjected to silica gel CC eluting with a gradient of CHCl3-acetone (4:1 to 0:1) to afford six sub-fractions (E4.1 to E4.6). Compounds 3 (15.8 mg) and 4 (16.2 mg) were obtained from sub-fraction E4.3 (250 mg) by using C18-RP silica gel CC and eluting with a gradient of MeOH–H2O (1:2 to 2:1). Fraction E5 (6.5 g) was also subjected to silica gel CC eluting with a gradient of CHCl3–MeOH (5:1 to 0:1) to afford eight subfractions (E5.1 to E5.8). The sub-fraction E5.6 (320 mg) was further subjected to C18-RP silica gel CC, eluted with a gradient of MeOH–H2O (1:3 to 1:1) to afford compound 5 (10.2 mg).
Trans-resveratrol (1)
Ivory amorphous powder; 1H-NMR (500 MHz, Methanol-d4) δH (ppm): 6.47 (2H, d, J = 2.0 Hz, H-2/H-6), 6.18 (1H, t, J = 2.0 Hz, H-4), 7.36 (2H, d, J = 8.5 Hz, H-2′/H-6′), 6.78 (2H, d, J = 8.5 Hz, H-3′/H-5′), 6.97 (1H, d, J = 16.0 Hz, H-8), 6.81 (1H, d, J = 16.0 Hz, H-7); 13C-NMR (125 MHz, Methanol-d4) δC (ppm): 159.6 (C-3/C-5), 158.3 (C-4), 141.3 (C-1), 130.4 (C-1′), 129.3 (C-8), 128.7 (C-2′/C-6′), 127.0 (C-7), 116.4 (C-3′/C-5′), 105.7 (C-2/C-6), 102.6 (C-4); ESI-MS m/z 229.09 [M+H]+ (Calcd. for C14H12O3).
Trans-piceatannol (2)
Ivory amorphous powder; 1H-NMR (500 MHz, Acetone-d6) δH (ppm): 8.36 (2H, s, 3/5-OH), 8.16 (1H, br s, 4′-OH), 8.06 (1H, br s, 3′-OH), 7.07 (1H, d, J = 2.0 Hz, H-2′), 6.93 (1H, d, J = 16.5 Hz, H-8), 6.89 (1H, dd, J = 8.5, 2.0 Hz, H-6′), 6.82 (1H, d, J = 8.5 Hz, H-5′), 6.81 (1H, d, J = 16.5 Hz, H-7), 6.53 (2H, d, J = 2.0 Hz, H-2/6), 6.25 (1H, s, H-4); 13C-NMR (125 MHz, Acetone-d6) δC (ppm): 159.5 (C-3/C-5), 146.0 (C-3′/C-4′), 140.7 (C-1), 130.5 (C-1′), 129.2 (C-8), 126.8 (C-7), 119.8 (C-6′), 116.1 (C-5′), 113.7 (C-2′), 105.5 (C-2/C-6), 102.6 (C-4); ESI-MS m/z 245.08 [M+H]+ (Calcd. for C14H12O4).
Sulfuretin (3)
Yellow amorphous solid; 1H-NMR (500 MHz, Methanol-d4) δH (ppm): 7.63 (1H, d, J = 8.5 Hz, H-5), 7.54 (1H, d, J = 2.0 Hz, H-8), 7.26 (1H, dd, J = 8.5, 2.0 Hz, H-6), 6.86 (1H, d, J = 8.0 Hz, H-5′), 6.73 (1H, dd, J = 8.0, 2.0 Hz, H-6′), 6.72 (1H, d, J = 2.0 Hz, H-2′), 6.71 (1H, s, H-2); 13C-NMR (125 MHz, Methanol-d4) δC (ppm): 184.5 (C-4), 169.8 (C-7), 168.4 (C-9), 149.3 (C-4′), 147.7 (C-3), 146.7 (C-3′), 126.8 (C-5), 126.3 (C-6′), 125.5 (C-1′), 118.9 (C-2′), 116.7 (C-5′), 114.8 (C-10), 114.6 (C-6), 114.1 (C-2), 99.3 (C-8); ESI-MS m/z: 271.06 [M+H]+ (Calcd. for C15H10O5).
(+)-Balanophonin (4)
Yellow amorphous powder;  +16.3° (c 0.05, MeOH); 1H-NMR (500MHz, Methanol-d4) δH: 9.60 (1H, d, J = 8.0 Hz, H-9′), 7.61 (1H, d, J = 15.5 Hz, H-7′), 7.31 (1H, s, H-6′), 7.25 (1H, s, H-2′), 6.97 (1H, s, H-2), 6.85 (1H, d, J = 8.0 Hz, H-5), 6.80 (1H, d, J = 8.0, H-6), 6.70 (1H, dd, J = 15.5, 8.0 Hz, H-8′), 5.63 (1H, d, J = 6.5 Hz, H-7), 3.92 (3H, s, 3′-OCH3), 3.91-3.87 (2H, m, H-9), 3.84 (3H, s, 3-OCH3), 3.59 (1H, q, J = 6.5 Hz, H-8); 13C-NMR (125 MHz, Methanol-d4) δC (ppm): 196.1 (C-9′), 156.8 (C-4′), 156.1 (C-3′), 152.9 (C-4), 149.2 (C-3), 146.0 (C-7′), 133.9 (C-1), 131.3 (C-5), 129.6 (C-1′), 127.1 (C-8′), 120.0 (C-6), 119.8 (C-6′), 116.2 (C-5), 114.3 (C-2′), 110.6 (C-2), 90.1 (C-7), 64.5 (C-9), 56.8 (3-OCH3), 56.4 (3′-OCH3), 54.6 (C-8); ESI-MS m/z: 357.14 [M+H]+ (Calcd. for C20H20O6).
+16.3° (c 0.05, MeOH); 1H-NMR (500MHz, Methanol-d4) δH: 9.60 (1H, d, J = 8.0 Hz, H-9′), 7.61 (1H, d, J = 15.5 Hz, H-7′), 7.31 (1H, s, H-6′), 7.25 (1H, s, H-2′), 6.97 (1H, s, H-2), 6.85 (1H, d, J = 8.0 Hz, H-5), 6.80 (1H, d, J = 8.0, H-6), 6.70 (1H, dd, J = 15.5, 8.0 Hz, H-8′), 5.63 (1H, d, J = 6.5 Hz, H-7), 3.92 (3H, s, 3′-OCH3), 3.91-3.87 (2H, m, H-9), 3.84 (3H, s, 3-OCH3), 3.59 (1H, q, J = 6.5 Hz, H-8); 13C-NMR (125 MHz, Methanol-d4) δC (ppm): 196.1 (C-9′), 156.8 (C-4′), 156.1 (C-3′), 152.9 (C-4), 149.2 (C-3), 146.0 (C-7′), 133.9 (C-1), 131.3 (C-5), 129.6 (C-1′), 127.1 (C-8′), 120.0 (C-6), 119.8 (C-6′), 116.2 (C-5), 114.3 (C-2′), 110.6 (C-2), 90.1 (C-7), 64.5 (C-9), 56.8 (3-OCH3), 56.4 (3′-OCH3), 54.6 (C-8); ESI-MS m/z: 357.14 [M+H]+ (Calcd. for C20H20O6).
+16.3° (c 0.05, MeOH); 1H-NMR (500MHz, Methanol-d4) δH: 9.60 (1H, d, J = 8.0 Hz, H-9′), 7.61 (1H, d, J = 15.5 Hz, H-7′), 7.31 (1H, s, H-6′), 7.25 (1H, s, H-2′), 6.97 (1H, s, H-2), 6.85 (1H, d, J = 8.0 Hz, H-5), 6.80 (1H, d, J = 8.0, H-6), 6.70 (1H, dd, J = 15.5, 8.0 Hz, H-8′), 5.63 (1H, d, J = 6.5 Hz, H-7), 3.92 (3H, s, 3′-OCH3), 3.91-3.87 (2H, m, H-9), 3.84 (3H, s, 3-OCH3), 3.59 (1H, q, J = 6.5 Hz, H-8); 13C-NMR (125 MHz, Methanol-d4) δC (ppm): 196.1 (C-9′), 156.8 (C-4′), 156.1 (C-3′), 152.9 (C-4), 149.2 (C-3), 146.0 (C-7′), 133.9 (C-1), 131.3 (C-5), 129.6 (C-1′), 127.1 (C-8′), 120.0 (C-6), 119.8 (C-6′), 116.2 (C-5), 114.3 (C-2′), 110.6 (C-2), 90.1 (C-7), 64.5 (C-9), 56.8 (3-OCH3), 56.4 (3′-OCH3), 54.6 (C-8); ESI-MS m/z: 357.14 [M+H]+ (Calcd. for C20H20O6).Cassigarol E (5)
Brown amorphous powder; −56.4° (c 0.12, MeOH); 1H-NMR (500 MHz, Methanol-d4) δH (ppm): 7.15 (1H, d, J = 2.0 Hz, H-2), 7.09 (1H, dd, J = 8.5, 2.0 Hz, H-6), 6.99 (1H, d, J = 16.0 Hz, H-7), 6.96 (1H, d, J = 8.5 Hz, H-5), 6.88 (1H, d, J = 16.0 Hz, H-8), 6.68 (1H, d, J = 2.0 Hz, H-2′), 6.67 (1H, d, J = 8.5 Hz, H-5′), 6.50 (1H, dd, J = 8.5, 2.0 Hz, H-6′), 6.48 (2H, overlap, H-12/H-12′), 6.19 (1H, t, J = 2.0 Hz, H-14′), 6.17 (1H, t, J = 2.0 Hz, H-10′), 6.12 (2H, d, J = 2.0 Hz, H-10/H-14), 4.75 (2H, d, J = 2.5 Hz, H-7′/H-8′); 13C-NMR (125 MHz, Methanol-d4) δC (ppm): 159.6 (C-11/C-11′), 159.2 (C-13/C-13′), 146.6 (C-4′), 146.1 (C-3′), 145.4 (C-4), 145.0 (C-3), 141.0 (C-9), 140.1 (C-9′), 132.6 (C-1), 129.4 (C-7), 129.0 (C-1′), 128.4 (C-8), 121.0 (C-6′), 120.7 (C-6), 118.1 (C-5), 115.9 (C-2), 115.8 (C-5′), 115.6 (C-2′), 107.4 (C-10′/C-14′), 105.9 (C-10/C-14), 103.6 (C-12′), 102.9 (C-12), 82.2 (C-8′), 81.8 (C-7′); ESI-MS m/z: 487.13 [M+H]+ (Calcd. for C28H22O8).
−56.4° (c 0.12, MeOH); 1H-NMR (500 MHz, Methanol-d4) δH (ppm): 7.15 (1H, d, J = 2.0 Hz, H-2), 7.09 (1H, dd, J = 8.5, 2.0 Hz, H-6), 6.99 (1H, d, J = 16.0 Hz, H-7), 6.96 (1H, d, J = 8.5 Hz, H-5), 6.88 (1H, d, J = 16.0 Hz, H-8), 6.68 (1H, d, J = 2.0 Hz, H-2′), 6.67 (1H, d, J = 8.5 Hz, H-5′), 6.50 (1H, dd, J = 8.5, 2.0 Hz, H-6′), 6.48 (2H, overlap, H-12/H-12′), 6.19 (1H, t, J = 2.0 Hz, H-14′), 6.17 (1H, t, J = 2.0 Hz, H-10′), 6.12 (2H, d, J = 2.0 Hz, H-10/H-14), 4.75 (2H, d, J = 2.5 Hz, H-7′/H-8′); 13C-NMR (125 MHz, Methanol-d4) δC (ppm): 159.6 (C-11/C-11′), 159.2 (C-13/C-13′), 146.6 (C-4′), 146.1 (C-3′), 145.4 (C-4), 145.0 (C-3), 141.0 (C-9), 140.1 (C-9′), 132.6 (C-1), 129.4 (C-7), 129.0 (C-1′), 128.4 (C-8), 121.0 (C-6′), 120.7 (C-6), 118.1 (C-5), 115.9 (C-2), 115.8 (C-5′), 115.6 (C-2′), 107.4 (C-10′/C-14′), 105.9 (C-10/C-14), 103.6 (C-12′), 102.9 (C-12), 82.2 (C-8′), 81.8 (C-7′); ESI-MS m/z: 487.13 [M+H]+ (Calcd. for C28H22O8).sEH Inhibitory Activity Assay
The sEH assay was performed as described previously.811 Briefly, 130 µL of sEH in 25.0 mM Bis-Tris-HCl buffer (pH 7.0) and 20.0 µL of the compounds (1 – 0.06 mM concentration) diluted in methanol, were added in 96-well plate, to which 50.0 µL of 20.0 µM PHOME was added in the mixture. After initiating the enzyme reaction at 37℃, the products by hydrolysis of the substrate were monitored at excitation and emission of 330 and 465 nm for one hour.
where C40 and S40 were the fluorescence of the control and inhibitor, respectively, after 40 min, S0 and C0 is the fluorescence of inhibitor and control, respectively, at 0 min. 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA) was used as a positive control.
Result and Discussion
After removing the oil (vegetable oil) from the seeds of P. edulis by n-hexane, the residue was extracted with ethanol to obtain ethanol extract. In the search for sEH inhibitors from natural sources, we found that the ethanol extract of the seeds of P. edulis inhibited 64.7% of sEH activity at a concentration of 37.5 µg/mL. This extract was then partitioned with dichloromethane (CH2Cl2), ethyl acetate (EtOAc) fractions and aqueous residue. In the preliminary experiment, we tested the inhibitory activity of these fractions at 37.5, 75.0 and 150.0 µg/mL.
Based on the results in Table 1, CH2Cl2- soluble fraction showed 91.8% inhibition at the concentration of 37.5 µg/mL, which was approximately 2.0-fold more potent than aqueous layer (48.7%). Interestingly, the EtOAc-soluble fraction exhibited very potently with > 100% sEH activity at the same concentration. Considering that EtOAc-soluble fraction showed the strongest action, our subsequent studies focused on the isolation of active components. This sub-fraction was subjected to column chromatography on a silica gel and C18-RP silica gel column to obtain five compounds (1 – 5) (Fig. 1).
Compound 1 was obtained as an ivory amorphous powder. The 1H-NMR of 1 showed signals the presence of 1,3,5-trisubstituted benzene ring characterized with AB2 system [δH 6.47 (2H, d, J = 2.0 Hz, H-2/H-6) and 6.18 (1H, t, J = 2.0 Hz, H-4)], and a 1,4-disubstituted benzene ring characterized with an A2B2 system [δH 7.36 (2H, d, J = 8.5 Hz, H-2′/H-6′) and 6.78 (2H, d, J = 8.5 Hz, H-3′/H-5′) together with trans-olefinic protons [δH 6.97 (1H, d, J = 16.0 Hz, H-8) and 6.81 (1H, d, J = 16.0 Hz, H-7)] (Fig. 1). The 13C-NMR spectrum of 1 exhibited 14 carbon signals including 12 aromatic carbons [δC 102.6 – 159.6], belonging to two benzene rings, and two olefinic carbons [δC 129.3 (C-7), and 128.7 (C-8)] (Fig. 1). The ESI-MS data of 1 indicated the pseudo molecular ion at m/z 229.09 for the [M+H]+, indicating a molecular weight of 228. Its molecular formula was determined to be C14H12O3, according to ESI-MS, 1H- and 13C-NMR spectroscopic data. Based on the above evidence and comparison with reported data,26 compound 1 was identified as trans-resveratrol. Compound 2, a derivative of 1, which the 1H- and 13C-NMR spectra were similar to those of 1 except for the presence of 1,3,4-trisubstituted benzene ring characterized with ABX system [δH 7.07 (1H, d, J = 2.0 Hz, H-2′), 6.89 (1H, dd, J = 8.5, 2.0 Hz, H-6′), and 6.82 (1H, d, J = 8.5 Hz, H-5′)] in 2 (Fig. 1). The ESI-MS spectrum of compound 2 showed the pseudo molecular ion at m/z 245.08 [M+H]+, indicating the molecular formula C14H12O4. Thus, compound 2 was identified as trans-piceatannol in comparison with literature data.27 These compounds (1 – 2) possess the basics of stilbene skeleton and are known to inhibit anti-oxidant, anti-inflammatory, anti-diabetes and anticancer.28
Compound 3 was obtained as yellow amorphous solid. The 1H-NMR of 3 showed signals of 1,2,4-trisubstituted benzene rings characterized with two ABX systems [δH 7.63 (1H, d, J = 8.5 Hz, H-5), 7.54 (1H, d, J = 2.0 Hz, H-8), and 7.26 (1H, dd, J = 8.5, 2.0 Hz, H-6); δH 6.86 (1H, d, J = 8.5, Hz, H-5′), 6.73 (1H, d, J = 2.0 Hz, H-2′), and 6.72 (1H, dd, J = 8.5, 2.0 Hz, H-6′)], together with an olefinic proton at δH 6.71 (1H, s, H-2) (Fig. 1). The 13C-NMR and distortionless enhancement by polarization transfer (DEPT) spectra of 3 showed signals for six aromatic quaternary carbon [δC 169.8 (C-7), 168.4 (C-9), 149.3 (C-4′), 146.7 (C-3′), 125.5 (C-1′), 114.8 (C-10)] and six aromatic carbons [δC 126.8 (C-5), 126.3 (C-6′), 118.9 (C-2′), 116.7 (C-5′), 114.6 (C-6) and 99.3 (C-8)] (Fig. 1). Furthermore, a carbonyl carbon at δC 184.5 (C-4), together with two olefinic carbons [δC 147.7 (C-3), and 114.1 (C-2)] were also observed in the 13C NMR spectrum indicated that 3 was aurone skeleton (Fig. 1).29 The ESI-MS spectrum of compound 3 showed the pseudo molecular ion at m/z 271.06 [M+H]+, indicating the molecular formula C15H10O5. Based on the above evidence and comparison with reported data,30 compound 3 was identified as sulfuretin, which was isolated from P. edulis for the first time. This compound possessed anti-inflammatory,31 anticancer,32 and neuroprotective activities.33
Compound 4 was isolated as yellow amorphous powder. The 1H-NMR spectrum of 4 showed an aldehyde proton at δH 9.60 (1H, d, J = 8.0 Hz, H-9′), trans-olefinic protons at δH 6.70 (1H, dd, J = 15.5, 8.0 Hz, H-8′) and 7.61 (1H, d, J = 15.5 Hz, H-7′), which were assigned to a trans-cinnamaldehyde moiety. In addition, the 1H-NMR of 4 showed two sets of aromatic protons signal [δH 6.97 (1H, s, H-2), 6.85 (1H, d, J = 8.0 Hz, H-5), 6.80 (1H, d, J = 8.0, H-6); δH 7.31 (1H, s, H-6′), 7.25 (1H, s, H-2′) arising from 1,3,4-trisubstituted and 1,3,4,5-tetrasubstituted benzene rings, respectively (Fig. 1). A stereochemistry of the dihydrobenzofuran ring [δH 5.63 (1H, d, J = 6.5 Hz, H-7), δH 3.59 (1H, q, J = 6.5 Hz, H-8) and δH 3.91-3.87 (2H, m, H-9)], and two singlets of methoxy groups [δH 3.92 (3H, s, 3′-OCH3), and 3.84 (3H, s, 3-OCH3)] were also observed in the 1H-NMR spectrum. The large proton coupling constant between H-7 and H-8 (J7,8 = 6.5 Hz) suggested the dihydrofuran ring has a trans-configuration. The 13C-NMR and DEPT spectra of 4 revealed the presence of twelve aromatic carbons, two olefinic carbons, an aldehyde carbon [δC 196.1 (C-9′)], a hydroxymethyl carbon [δC 64.5 (C-9)] and two methoxy carbons [δC 56.8 (3-OCH3), and 56.4 (3′-OCH3)] (Fig. 1). The HMBC correlations of proton H-2 at δH 6.97 (1H, s) and methoxy protons at δH 3.84 (3H, s) to carbon signal at δC 149.2 (C-3), as well as proton H-2′ at δH 7.25 (1H, s) and methoxy protons at δH 3.92 (3H, s) to carbon signal at δC 156.1 (C-3′), suggested that two methoxy groups were located at C-3 and C-3′ (Fig. 1). Further analysis of these signals by the COSY, HMQC and HMBC spectra led to the partial structures of 4. The ESI-MS data of 4 indicated the pseudo molecular ion at m/z 357.14 for the [M+H]+, indicating a molecular weight of 356. Its molecular formula was determined to be C20H20O6, according to ESI-MS, 1H- and 13C-NMR spectroscopic data. Therefore, compound 4 was identified as (+)-balanophonin,34 which was isolated from Passiflora genus for the first time. This compound was known to possess anti-oxidant,3536 anti-cholinesterase, 36 anti-inflammatory, anticancer, and anti-neurodegenerative activities.37
Compound 5 was isolated as brown amorphous powder. The 1H-NMR spectrum of 5 showed the presence of two ABX system signals for the A ring [δH 6.68 (1H, d, J = 2.0 Hz, H-2′), 6.67 (1H, d, J = 8.5 Hz, H-5′), 6.50 (1H, dd, J = 8.5, 2.0 Hz, H-6′)] and the D ring [δH 7.15 (1H, d, J = 2.0 Hz, H-2), 7.09 (1H, dd, J = 8.5, 2.0 Hz, H-6), 6.96 (1H, d, J = 8.5 Hz, H-5)], two sets of AB2 system signals for the B and C rings [6.48 (2H, overlap, H-12/H-12′), 6.19 (1H, t, J = 2.0 Hz, H-14′), 6.17 (1H, t, J = 2.0 Hz, H-10′), 6.12 (2H, d, J = 2.0 Hz, H-10/H-14)], two doublets for the trans-olefinic protons [δH 6.99 (1H, d, J = 16.0 Hz, H-7), 6.88 (1H, d, J = 16.0 Hz, H-8)], and two equivalent oxybenzyl methine protons [δH 4.75 (2H, d, J = 2.5 Hz, H-7′/H-8′)] (Fig. 1). The 13C-NMR and DEPT spectra of 5 exhibited 28 carbon signals including 24 aromatic carbons [δC 102.9 – 159.6], belonging to four benzene rings, and two olefinic carbons [δC 129.4 (C-7), and 128.4 (C-8)]. In addition, the carbon signals [δC 82.2 (C-8′) and 81.8 (C-7′)] indicate the presence of two equivalent oxybenzyl methine carbons. Analysis of these signals by the COSY, HMQC and HMBC spectra led to the partial structures of compound 5 (Fig. 1). The ESI-MS data of 5 indicated the pseudo molecular ion at m/z 487.13 for the [M+H]+, indicating a molecular weight of 486. Its molecular formula was determined to be C28H22O8, according to ESI-MS, 1H- and 13C-NMR spectroscopic data. The relative configuration between the C-7′ and the C-8′ positions was concluded to be cis from the coupling constant of the two oxybenzylmethine protons (J = 2.5 Hz). Based on the above evidence and comparison with reported data,38 compound 5 was identified as cassigarol E, which was isolated from Passiflora genus for the first time. Cassigarol E possessed anti-HIV-1,39 antidiabetic,40 and anticancer activities.41
The effect of isolated compounds (1 – 5) from P. edulis on sEH inhibitory activities was evaluated. The sEH inhibitory activities were determined using recombinant human sEH incubated with PHOME, an artificial substrate for fluorescence detection with AUDA (IC50 value 4.4 nM), a sEH inhibitor as the positive control. The result showed that compound 4 had no inhibitory (N.I) effect on the activity of enzyme sEH at a concentration of 100 µM, while compounds 1 and 2 at a concentration of 100 µM exhibited the highest sEH inhibitory activity (>100%) (Table 1). Sulfuretin (3), and cassigarol E (5) also showed a strong inhibitory effect on sEH with their inhibition values in the range of 74.6 and 77.7%, respectively (Table 1). Compounds 1 – 3, and 5 showed inhibitory rates over 50% and were further evaluated at concentrations ranging from 6.2 to 100 µM, to elucidate the IC50 values. These inhibitors (1 – 3, and 5) showed dose-dependent inhibition, with IC50 values of 14.2 ± 0.6, 3.4 ± 4.8, 15.8 ± 1.0, and 14.4 ± 0.8 µM, respectively (Table 2).
Previously, natural plant components with stilbene skeletons from other plants have also shown inhibitory effects on sEH activity. Rhapontigenin, isorhapontin and astringin from Rheum undulatum (Polygonaceae),8 and 2-isopropyl-5-[(E)-2- phenylvinyl]benzene-1,3-diol have displayed potent sEH inhibitory activity.42 Several stilbene from Polygonum multiflorum (Polygonaceae), such as (E)-2,3,5,4′-tetrahydroxystilbene-2-O-β-D-glucoside, (E)-2,3,5,4′-tetrahydroxystilbene-2-O-β-D-xyloside, and (E)-2,3,5,4′-tetrahydroxystilbene-2-O-β-D-(6′′-O-acetyl)-glucoside completely inhibited sEH in a dose-dependent manner, with low IC50 values.43 As in this study, some isolated stilbenes from P. edulis can manifest sEH inhibitory activity. Such compounds have been purified from natural medicinal plants for many years all over the world. In addition to other medicinal properties, such as anti-inflammatory, anticancer, and antioxidant activities, the sEH inhibitory activity is worthy of notice, because low-molecular-weight materials can easily reach the site of action following oral administration since they cross the blood-brain barrier.44
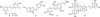


 XML Download
XML Download