

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Pancreatic cancer is the fourth leading cause of cancerrelated deaths at all ages. It has a 5-year survival rate of less than 7%.12 The mortality rate of pancreatic cancer is high, but its cause or underlying mechanisms are not fully understood.3 Surgical resection, chemotherapy, and radiotherapy were used as the main therapeutic options,45 with gemcitabine being the typical chemotherapeutic agent used for pancreatic cancer. However, the response rate to gemcitabine is less than 10%.6 Thus, novel agents to sensitize pancreatic cancer cells are needed.
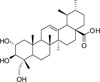
Asiatic acid is a pentacyclic triterpene isolated from the Centella asiatica, which is a plant used in traditional medicine in Asia.7 It displays antioxidant, anti-inflammatory, and neuroprotective properties.89 Several studies have shown that asiatic acid induces apoptosis in Hep G2 cells causing the release of Ca2+ and up-regulation of p53.10 Furthermore, asiatic acid can prevent the growth of HT-29 cells by inducing apoptosis and affecting B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma-extralarge (Bcl-xL) expression.11 It has also been reported that asiatic acid inhibits angiogenesis through VEGF and human gliomas in endothelial cells.12
As though the potential of asiatic acid in cancer, anti-proliferative effect of asiatic acid and the underlying mechanism(s) have not evaluated in pancreatic cancer. In this study, the cytotoxic effects of asiatic acid were investigated in pancreatic cancer cell lines and the molecular mechanisms were identified using the TUNEL assay, annexin V assay, western blotting, and miRNA analysis.
Experimental
General experimental procedure
Human pancreatic cancer cell lines, MIA PaCa-2 and PANC-1 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, US). MIA PaCa-2 and PANC-1 were cultured in Dulbecco's modified Eagle's medium (DMEM; Hyclone, Waltham, MA, USA) with 10% Fetal bovine serum (FBS; Hyclone, Waltham, MA, USA) and 1% penicillin/streptomycin (Gibco, Invitrogen Inc., Carlsbad, CA, USA) at 37 ℃ with 5% CO2. Asiatic acid was purchased from Sigma-Aldrich (St.Louis, MO, U.S.A.) with a purity of 97%.
Water soluble tetrazolium salt-1 (WST-1) assay
Asiatic acid is dissolved in dimethyl sulfoxide (DMSO) at 60 mM as a stock solution and stored at −20 ℃. The cancer cell lines were seeded in 100 µL of medium at 5 × 103 cells per well in 96-well plates. Cells were incubated for 24 h, then treated with asiatic acid at various concentrations (0, 20, 40 and 60 µM) and 0.1% (v/v) DMSO as a negative control. After 48 and 72 h, 10 µL of WST-1 (Roche, Mannheim, Germany) reagent was added to each well and incubated for 2 h at 37 ℃. Absorbance was measured at 450 nm using a microplate reader. Cell viability was calculated as a percent of the control value and mean values were averaged for three wells. This experiment was repeated three times.
Western blotting
MIA PaCa-2 cells (5 × 105 per 1 ml in 10 cm2 culture dish) were treated with asiatic acid at various concentrations (0, 20, 40, and 60 µM) for 48 h. Cells were treated with 1 mL radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific, Waltham, MA, USA) with added phosphatase inhibitor (Roche, Mannheim, Hermany) on ice for 30 min for lysis. The bicinchoninic acid (BCA) Protein Assay Kit (Thermo Scientific, USA) was used to measure total protein concentration compared to a protein standard; 30 µg of total protein was loaded on SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. After blocking in 5% nonfat dry milk in Tris buffered saline with Tween-20 (TBST) for 1 h, the membranes were incubated overnight at 4 ℃ with primary antibodies: anti-human-LC3II, mTOR, phosphomTOR, AMPK, phospho-AMPK, p38, phopho-p38, caspase-3, cleaved caspase-3, cleaved poly ADP ribose polymerase (PARP), and alpha-tubulin from Cell Signaling Technology (Beverly, Mass., USA), phosphatase and tensin homolog (PTEN) from Bethly Laboratories (Montgomery, TX, USA) and Bcl-2 from Enzo Life Sciences (Farmingdale, NY, USA). Next, membranes were incubated for 1 h with secondary antibodies: anti-rabbit from Thermo Scientific (Waltham, MA, USA), and anti-mouse from Bethly Laboratories (Montgomery, TX, USA). Protein bands were detected with an ECL solution kit purchased from Thermo Scientific (Waltham, Ma, USA) and the relative expression of protein bands was analyzed with the FluoroChem E image analyzer (Cell Bioscience, Santa Clara, CA, USA).
UNEL assay
MIA PaCa-2 cells (1.5 × 104 cells/200 µL) were seeded into a Chamber Slide (Lab Tek Chamber Slide) for 24 h. Cells were treated with asiatic acid at IC50 and HSW34513 at IC50 as a positive control for apoptosis. After incubation for 48 h, the cells were fixed with 100 µL of 4% paraformaldehyde, and then the TUNEL assay was performed using the In Situ Cell Death Detection Kit (Roche, Mannheim, Germany).
Annexin V assay
MIA PaCa-2 cells (4 × 105 cells/mL) were seeded onto 60 mm dishes. The following day, the cells were treated with asiatic acid at 0, 20, 40 and 60 µM for 48 h. DMSO was used as a negative control. After incubation, cells were fixed and stained using the Muse® Annexin V and Dead Cell Assay Kit (Merck Millipore, Billerica, USA) using the supplier's protocol.
Total RNA extraction
MIA PaCa-2 cells (4 × 105 cells/well) were seeded on 60 mm dishes. The following day, cells were treated with asiatic acid at 60 µM and incubated for 48 and 72 h. Cells were treated and incubated with DMSO as a negative control. After incubation, the total RNA was extracted using a Trizol reagent (Invitrogen, CA, USA) using the supplier's protocol.
TaqMan miRNA assay
To examine miRNA expression, the TaqMan miRNA assay was performed. A total 50 ng RNA was converted to cDNA using the TaqMan® microRNA Reverse Transcription Kit (Applied Biosystems) following the manufacturer's protocol. Real-time PCR was performed in triplicates and the 2−ΔΔCT method was used for the relative quantitation of samples. Data was normalized with the small nuclear RNA, RNU6B gene.
Result and Discussion
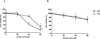
In order to investigate the cytotoxic effect of asiatic acid (Fig. 1) on pancreatic cancer cells, MIA PaCa-2 and PANC-1 cells were exposed to various concentrations of asiatic acid (0, 20, 40, and 60 µM) for 48 and 72 h (Fig. 2). Cell viability was analyzed using the WST-1 assay. The viability of MIA PaCa-2 cells decreased by 93.3, 84.6, and 30.2% of control levels at increasing doses after 48 h. The cell viability of PANC-1 cells decreased by 88.8, 85.5, and 68.3% of control levels at increasing doses after 48 h. The viability of MIA PaCa-2 cells was reduced by 41.8 and 13.6% of control levels at 40 and 60 µM, respectively, after 72 h. The viability of PANC-1 cells dropped by 93.7, 77.6, and 72.1% at increasing doses after 72 h. The viability of both cell lines decreased in a dose-dependent manner, most prominently at 60 µM. IC50 of asiatic acid in MIA PaCa-2 was 52.7 µM. As the cytotoxic effect was more evident in MIA PaCa-2 than in PANC-1 cells, subsequent experiments were performed with MIA PaCa-2 cells.
Tang et al. reported that asiatic acid induced cell growth inhibition and apoptosis through a mitochondrial death cascade in colon cancer.14 Wu et al. also found that asiatic acid induced apoptosis in HL-60 human leukemia cells by modulating the Bcl-2 family.15 The TUNEL assay is a method used for detecting DNA fragmentation that results from apoptosis.16 To confirm whether asiatic acid induces apoptosis in MIA PaCa-2 cells, the TUNEL assay, western blotting, and annexin V assay were performed. For the TUNEL assay, MIA PaCa-2 cells were treated with 52.7 µM of asiatic acid (IC50 value) and 5 µM HSW354 (IC50 value), a positive inducer of apoptosis.13 Similar to HSW345, asiatic acid induced apoptosis (Fig. 3A). Using western blots, it was found that asiatic acid treatment inhibited the expression of antiapoptotic Bcl-2 and resulted in the cleavage of caspase-3 and PARP. The expression of PTEN, a protein related to cell cycle control, was biphasically changed depending on concentration (Fig. 3B). The flow cytometry analysis of the cell cycle also showed no significant changes (data not shown). According to the annexin V assay results, apoptosis rates were increased by asiatic acid (Fig. 3C). Caspase proteins are major mediators of apoptosis and cleaved caspase-3 is a marker for apoptosis.17 These results confirm that asiatic acid induces apoptosis in MIA PaCa-2 cells.
To determine whether asiatic acid induces autophagy in pancreatic cancer cells, levels of autophagy protein markers were determined by using western blotting. Autophagy is the major pathway to eliminate damaged cell organelles or useless proteins. With regard to cancer, autophagy possesses two opposing functions.1819 Autophagy is a tumor suppressor mechanism, whereby the lack of autophagy genes has been shown to promote tumorigenesis.20 In contrast, autophagy can contribute to tumor growth by protecting cells from anticancer agents.212223 LC3I is cleaved to form LC3II, which is involved in the formation of autophagosomes; LC3II is considered a marker of autophagy. p38 is a Mitogen-activated protein kinase (MAPK) family member and has been reported to be related to the induction of autophagy.2425 Choi et al. reported that capsaicin-induced p-p38 led to autophagy and regulated autophagosome formation in breast cancer cells.26 Decreased mTOR signaling is an important initiator of autophagy to protect the cell from stresses, such as nutrient starvation and low ATP or oxygen levels. The inhibition of mTOR is known to induce early activation of the autophagy cascade, including the formation of autophagosomes.2728 Under insufficient ATP conditions, changes to the intracellular ATP/AMP ratio activates AMPK and phosphorylates tuberous sclerosis complex 2 (TSC2), which results in the inhibition of mTOR-mediated TSC2.29 The protein levels of LC3II, p38, p-p38, mTOR, p-mTOR, AMPK, and p-AMPK were examined (Fig. 4). Asiatic acid increased the level of LC3II, a marker of autophagosome formation, as well as the level of p-p38. In addition, the levels of p-mTOR, a negative regulator of autophagy, were decreased by asiatic acid. By contrast, the level of p-AMPK, a positive regulator of autophagy, was increased. Therefore, asiatic acid induced autophagy in MIA PaCa-2 cells through the AMPK/mTOR pathway.
miRNAs are small, non-coding RNA that play important roles in growth, differentiation, and cell death.30 Several miRNAs are differentially expressed in pancreatic cancer. Among them, miR-17 and miR-21 have been reported to be overexpressed in pancreatic cancer. miR-21 has been shown to be overexpressed in early pancreatic tumors and pancreatic cancer-derived cell lines associated with a poor outcome. miR-21 protects pancreatic cancer cells from cell death and inhibition of miR-21 suppresses tumor growth.31 miR-17 is a member of the miR-17-92 cluster, which is known to act as an oncogene in various cancers, such as breast, colon, lung, and pancreatic cancer.3233 Up-regulated miR-17 promotes pancreatic cancer cell proliferation and invasion.34 Using the TaqMan miRNA assay, we found that the expression levels of both miR-17 and miR-21 (Fig. 5A, 5B) were decreased compared to the levels in controls. Asiatic acid significantly reduced the overexpression of miR-17 and miR-21 in pancreatic cancer cells.
Therefore, asiatic acid induces apoptosis and autophagy in pancreatic cancer cell lines and asiatic acid may have potential to be developed as a therapeutic agent for pancreatic cancer.



 XML Download
XML Download