

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acromegaly is a rare disease. Its prevalence is 2.8–13.7 cases per million and incidence is 0.2–1.1 cases per million per year (1). It results from excess of the growth hormone (GH) and is mostly related with pituitary adenoma (12). Cardiovascular complications including cardiomyopathy are a major cause of morbidity and mortality in patients with acromegaly (2345). Currently, cardiac magnetic resonance imaging (CMR), especially advanced CMR techniques including T1 mapping and extracellular volume fraction (ECV) values play a key role in diagnosis of cardiomyopathies and prediction of the prognosis (67). However, CMR findings have been rarely described in acromegaly patients. We report a case of acromegaly cardiomyopathy presenting as dilated cardiomyopathy (DCM) with systolic dysfunction.
CASE REPORT
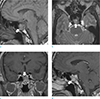
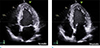
An age 38 male with history of hypertension was referred to the Department of Endocrinology with worsening headache. He noticed progressive enlargement of his face, hands, and feet for approximately 10 years and could not wear a hat as well as gloves that used to fit. Physical examination revealed frontal bossing, mandibular enlargement with prognathism, and widened space between the lower incisor teeth. He also had deep, husky voice, which he previously did not have. His initial blood pressure and body temperature were 160/104 mmHg and 37.1℃, respectively. Laboratory investigation showed the following results: random serum GH, 23.33 ng/mL (normal range, 0.1–1.5 ng/mL); insulin-like growth factor 1 (IGF-1), 592 ng/mL (normal range, 177–382 ng/mL); thyroid stimulating hormone (TSH), 0.66 uIU/mL (normal range, 0.4–4.1 uIU/mL); cortisol, 17.4 ug/mL (normal range, 5–25 ug/mL); and prolactin, 7.4 ng/mL (normal range, 0–25 ng/mL). GH failed to suppress during an oral glucose tolerance test (OGTT; nadir serum GH, 13.96 ng/mL [normal range, < 1 ng/mL]). MRI of the brain revealed a 20 mm mass at the pituitary gland (Fig. 1). Considering his clinical history and laboratory and image findings, acromegaly caused by the GH secreting pituitary macroadenoma was the most likely diagnosis. Surgical removal was planned for the pituitary mass and electrocardiogram (ECG) abnormalities were found during preoperative evaluation. An ECG showed a sinus rhythm with a heart rate of 61 beats per minute (bpm), right bundle branch block, T wave abnormality, and moderate voltage criteria for left ventricular hypertrophy (LVH), which may be a normal variant. Echocardiography revealed dilated left ventricle (LV) with global mild hypokinesia with mild LV systolic dysfunction (calculated ejection fraction [EF]: 49%) (Fig. 2). To evaluate the possibility of cardiomyopathy, CMR was recommended.
CMR was performed using a 3-T MR scanner (Skyra 3T, Siemens Healthcare, Erlangen, Germany) with ECG gating. As for findings of echocardiography, there were dilated LV showing increased end-diastolic volume of 153.9 mL/m2 and global hypokinesia. The right ventricular (RV) morphology and regional wall motion were normal, and pericardial effusion was not observed. The left-ventricular indexed systolic function was reduced (LVEF, 44.8%). Contrary to the echocardiographic findings, there was no evidence of mitral regurgitation in cine imaging.
Late gadolinium enhancement (LGE) images were taken 15 minutes after contrast administration. There was mid-layer patchy LGE in the mid inferoseptal LV wall (Fig. 3). Modified Look-Locker Inversion Recovery (MOLLI) scheme was used for native T1 mapping, using two inversions to acquire eight images over 11 heartbeats. The native T1 value was 1280 msec at the mid inferoseptal wall where positive LGE and 1250 msec at the mid anteroseptal wall, where negative LGE respectively (reference value, 1052 ± 23 msec at 3 T). ECV fraction was 28.9% (reference value, 0.26 ± 0.04% at 3T), calculated at the mid anteroseptal wall, where LGE negative myocardium (6).
The patient underwent trans-sphenoidal excision of the pituitary mass and biopsy revealed pituitary adenoma. Finally, the patient was diagnosed with acromegaly secondary due to GH secreting pituitary adenoma. He is on follow-up with medication for hypertension and cardiomyopathy.
DISCUSSION
Acromegaly is a relatively rare condition caused by excessive production of GH produced by pituitary adenoma beginning in the somatotropin cells. The median age at diagnosis is in the fifth decade of life and the slow progression of acromegaly leads to a diagnostic delay of average 4.5–5 years (1). Elevated GH increased serum IGF-1 by stimulating IGF-1 synthesis at the liver and pancreas. The representative clinical findings are acral and soft tissue overgrowth, joint pain, diabetes mellitus, hypertension, and heart and respiratory failure (2). Among them, cardiovascular involvement is one of the most severe complications of acromegaly, contributing significantly to mortality in this disease. Exposure to elevated GH and IGF-1 cause major structural and functional cardiac changes by affecting myocyte growth and structure, cardiac contractility, and vascular function (23458).
Acromegalic cardiomyopathy is the generic term for cardiac complications associated with acromegaly and involves nearly all aspects of the cardiovascular system: systolic and diastolic ventricular function, valvular heart disease, arrhythmia and eventually causing congestive heart failure (CHF) (2345). In the early phase of acromegaly, it manifests commonly as hyperkinetic syndrome, characterized by increased heart rate and increased systolic output (245). If the GH/IGF-I excess is left untreated, myofibrillolysis and fibrosis occur, developing diastolic dysfunction and eventually impairing the systolic function ending in heart failure (2345). Bihan et al. (3) revealed CHF is a very rare complication of acromegaly, occurring in less than 3% of patients. Among them, an acute episode due to hypertensive crisis or arrhythmia tends to be reversible with rapid recovery of myocardial dysfunction. However, in advanced stage, ventricular dilatation, arrhythmia and valvular disease may be irreversible (3).
CMR findings of acromegalic cardiomyopathy appear to vary from the early stage to the late stage (23458). LVH and myocardial fibrosis are common findings of acromegalic cardiomyopathy (4). Frequency of LVH detected on CMR varied from 5–72% (4) and DCM is a more rare complication (38). In our case, CMR showed dilated LV with decreased systolic function. Considering that the patient's symptoms have existed for a lengthy period, these findings probably correspond to the advanced stage of acromegalic cardiomyopathy. Unlike LV wall thinning in idiopathic DCM, LV wall thickness was relatively preserved in our case. This finding may be because ventricular hypertrophy occurs first before the development of DCM.
Biventricular hypertrophy is a significant pathognomonic feature of acromegalic cardiomyopathy (5). However, there is controversy regarding the impact of GH and IGF-1 relative to the RV. A study conducted by a Polish health center reported that there was no difference in the RV size, and thickness of the RV free wall among acromegaly patients and the control group (5). The morphology and function of the RV was also preserved in our patients. Further large population studies are needed on the effect to the RV.
In a study of CMR findings in patients with acromegaly, approximately 13.5% (5/40) of patients have myocardial LGE with various LGE pattern. One of them showed extensive mesocardial distributed LGE, another showed transmural LGE and the others (3/5) showed a small area of mesocardial LGE (4). In our patient, patchy with longitudinal mid wall LGE was present in the mid-inferoseptal wall, which did not correspond to any particular coronary artery distribution, consistent with a non-ischemic cardiomyopathy. However, due to the small numbers of cases, there was no study on the effect of LGE distribution and extent on cardiac prognosis on the patient with acromegaly.
ECV is a marker of myocardial tissue remodeling and provides a physiologically intuitive unit of measurement. Increased ECV is due to collagen deposition at interstitial space except for the cardiac amyloidosis (6). In our case, calculated ECV fraction was 28.9% showing upper normal limits. ECV value in acromegaly patients are thought to be affected by the degree of exposure to GH and IGF-1, and extent of myocyte hypertrophy, and interstitial fibrosis. In the study mentioned above, among the five patients who showed LGE, only one patient showed increased ECV fraction (4). It seems that the degree of interstitial fibrosis and degree of myocyte hypertrophy seem to conflict with ECV.
Main differential diagnosis is hypertensive cardiomyopathy. Hypertensive cardiomyopathy shows symmetric concentric hypertrophy of LV with maximum wall thickness usually involving the septum with diastolic or systolic dysfunction due to persistent systemic hypertension (9). In hypertensive cardiomyopathy, patchy LGE was found in approximate 50% of patients with LVH due to arterial hypertension (9). Additionally, acromegaly patients often have hypertension, and thus, it is difficult to differentiate between hypertensive cardiomyopathy and acromegalic cardiomyopathy using imaging.
In conclusion, we report a rare case of acromegalic cardiomyopathy, caused by GH secreting pituitary macroadenoma, which manifests DCM showing patchy LGE on CMR. Because CMR is increasingly used to differentiate the various etiology of cardiomyopathies, radiologists should be familiar with findings of CMR in patients with acromegaly.

 XML Download
XML Download