

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cardiovascular disease (CVD) is a leading cause of mortality and morbidity worldwide, contributing to over 17.6 million deaths annually.1) However, while the proportion of CVD cases has grown by approximately 14.5% globally over the period 2006–2016, this does not reflect a uniform increase in CVD cases geographically. For example, in contrast to the global increase in CVD cases, Organisation for Economic Co-operation and Development (OECD) member countries have shown an average reduction in CVD related mortality by approximately 42% from 1985–2005.2) Korea specifically has shown a faster decrease in CVD mortality than the OECD average at 182 cases per 100,000 population.2) Many explanations for diverging trends in CVD incidence exist, ranging from differing intervention strategies on modifiable risk factors such as smoking, to differing country-specific configurations of non-modifiable risk factors, such as age or genetic predisposition. These patterns have served to motivate CVD research, with the primary aim of identifying novel and effective targets for the clinical treatment and prevention of CVD.
The proliferation of studies including large-scale genetic data has had a profound impact upon CVD related research. Through genome-wide association study (GWAS), conducted with increasingly large sample sizes, it has been possible to identify candidate genetic variants associated with CVD related illness. Further, the emergence of Mendelian randomization (MR) approaches has presented the possibility for causal relationships to be estimated whilst controlling for residual confounding, a persistent concern in observational analyses. These developments have, in tandem, allowed for novel genetic drug targets to be identified and previously identified risk factors to be re-evaluated from differing perspectives. Genetic studies can be valuable in identifying groups predisposed to health outcomes, whilst MR focuses on estimating the causal contribution of life-long exposure to modifiable risk factors.
In many cases studies aim to elucidate the causal mechanisms underlying CVD, highlighting key aspects of the aetiology of CVD related illness which are suitable for intervention. However, owing to strengths and limitations unique to a given study design, it is often difficult to infer causality based on a single study. This motivates the triangulation-based approach to evidence evaluation, proposed by Lawlor et al.,3) which focuses upon agreement in research findings across studies of varying design. If each study design is subject to differing and unrelated forms of potential bias, substantial agreement in research findings across multiple different studies limits the extent to which findings are the result of bias specific to study design.3) Alternatively, observed differences in research findings can indicate the presence of one or more study-specific features which introduce bias into estimates of association.
In this review we critically evaluate the contribution of studies using observational, genetic, and MR study designs within a triangulation framework, exploring the extent to which patterns are observable with respect to identified risk factors for CVD. In particular, we focus upon the role of MR as a novel source of evidence in CVD research and highlight recent developments in the MR literature which warrant further attention. Finally, we link identified risk factors to the development of precision medicine and novel interventions, highlighting key areas which warrant attention in future CVD research.
CARDIOVASCULAR DISEASE AND THE KOREAN POPULATION
CVD represents a range of illnesses related to cardiovascular health including, but not limited to, hypertension, coronary heart disease (CHD), myocardial infarction (MI), heart failure, and stroke. It has been estimated that approximately 80% of CVD cases are preventable, and a wide range of potential modifiable risk factors for CVD have been previously identified.4) Historically, studies identifying CVD related risk factors have utilised samples of European ancestry, making it difficult to generalise research findings internationally. However, the emergence of large-scale cohort studies in non-European populations provides an opportunity for replication in populations of differing ancestry. One such study is the Korean Cancer Prevention Study-II (KCPS-II) Biobank, which as a blood-based cohort with long-term follow-up provides valuable information with respect to modifiable risk factors and genetic determinants of CVD.5)
The Korean Cancer Prevention Study-II Biobank
KCPS-II Biobank is a large blood-based cohort study comprised of 156,701 participants (94,840 men and 61,861 women) recruited from 18 health promotion centres across South Korea from 2004–2013.5) Participants underwent a health assessment including a self-complete questionnaire and medical examination, collecting data related to social status, health status, behaviour, physiology, and serum measures. Long-term follow-up; specifically, for cancer incidence, hospital admissions, and cause-specific mortality, was obtained using linkage to subsequent medical examinations at health promotion centres across South Korea. Further details available in a previously published cohort profile.5)
The inclusion of blood-based measures presents a valuable opportunity to elucidate the causal mechanisms behind CVD in the Korean population. Such measures allow for lipid concentrations, such as low-density lipoprotein cholesterol (LDL-C), to be examined within the context of CVD, and crucially, subsequent genotyping of KCPS-II Biobank participants allows for genetic determinants of CVD to be evaluated. The availability of genetic data in a non-European sample allows for candidate genetic variants implicated in CVD incidence to be re-evaluated and facilitates the application of MR approaches. Throughout this review, we relate key findings to the Korean population where available and highlight emerging research relevant to East Asia.
OBSERVATIONAL ANALYSES OF CARDIOVASCULAR DISEASE
Previous research has drawn attention to several risk factors for CVD related illness identified using observational analyses. Such studies include large-scale population based studies, in particular the Framingham Heart Study, the CArdiovascular research using LInked Bespoke studies and Electronic health Records (CALIBER) programme, and studies using Kaiser-Permanente data.6)7)8)9)10) Further details regarding each study are provided in the web appendix.
Lipids
There is a substantial body of research linking serum lipids to the onset of CVD.11)12)13) Observed changes in lipid levels arising from migration and interventions, and the subsequent impact on incidence of CVD initially suggested that total serum lipid levels could serve as an indicator of CVD risk.14)15) Subsequent observational analyses have shown evidence of a positive association between LDL-C and CVD.16)17)18) However, the relationship of high-density lipoprotein cholesterol (HDL-C) and triglycerides with respect to CVD remains controversial. Initially, HDL-C was found to be protective against CVD; however, subsequent studies have highlighted that this may not universally be the case.19)20) Rather, functionality of high-density lipoprotein may be more important, with high levels of HDL-C becoming pro-inflammatory only under specific conditions.21)22) Triglycerides have also been found to be a useful biomarker for CVD, but this could likely be due to their association with causal risk factors such as apo CIII.23)
These observed relationships between lipids and CVD have largely been replicated in East Asia and Korea specifically.14)24) Using data from KCPS-II Biobank Jung et al found LDL-C to be a strong predictor of stroke, warranting inclusion in a general risk score comprised of several traditional risk factors.25) Using a similar approach, Jee et al.26) found LDL-C to be highly predictive of CVD using data from the Korean Heart Study. Strong associations were also found for HDL-C and triglycerides, adjusting for LDL-C.
Alcohol consumption
Alcohol has been implicated as a risk factor for CVD related illness for decades. In 1974, Klatsky et al.27) identified a positive association between alcohol consumption and MI using data from the Kaiser Permanente Health Plan in California. This case-control study also found alcohol to be a risk factor independent of smoking status.27) However, whilst alcohol appears to be causally linked with many forms of CVD, the range of associations has been shown to be diverse across CVD related illness.28) Subsequent analyses using electronic health records from CALIBER have found heavy drinking to be associated with increased risk of coronary death and ischaemic stroke, whilst participants not consuming alcohol appear to be at greater risk for angina, MI, and ischaemic stroke.8) Such findings seem to suggest a protective effect of alcohol in low doses, though such an interpretation remains highly controversial.29)30)
Several explanations have been suggested for observed disagreement in research findings, such as in the case of MI. Studies using electronic health records often determine drinking status using proxy measures, and may lead to problems of misclassification.31) This differs markedly from the case-control design of the Kaiser Permanente Health Plan Study for example, which used questionnaires to determine levels of alcohol consumption.10)27) The divergence in association estimates with respect to alcohol and CVD related illness could also be due to residual confounding, whereby alcohol appears protective due to its correlation with an unobserved factor protective for CVD risk.
In the Korean population, several studies have identified alcohol as a risk factor for CVD related illness. Yoon et al.32) find alcohol to be positively associated with metabolic syndrome, and subsequent onset of CVD. Regarding the protective effect of mild alcohol consumption, a systematic review and meta-analysis conducted by Park et al found no evidence of an inverse association between alcohol and CVD in the Korean population.33)
Smoking
Smoking has been identified as a risk factor for CVD, with a positive relationship between the number of cigarettes smoked and CVD related illness.34)35) The effects of tobacco smoke have been shown to extend to second-hand smoking, whilst smoking cessation has been shown to substantially reduce CVD risk.34)36) Previous work stemming from the Korean Life Course Health Study and Korea Medical Insurance Corporation Study has identified smoking as a major independent risk factor for a range of CVD related illness, including ischaemic heart disease, CVD, and atherosclerotic CVD.37)38) Further, the effects of smoking on atherosclerotic CVD has been found to be independent of low-cholesterol levels.37) Further studies have echoed these findings within East Asia and Korea, finding strong evidence of a connection between smoking behaviour and CVD risk.39)40)
Obesity and C-reactive protein
Increases in adiposity, and obesity specifically, have been argued to be an independent risk factor for CVD and all-cause mortality.41) Using body mass index (BMI) as a measure of adiposity, it has been shown that higher levels of BMI increase risk of CVD related illness.42)43) Obesity has been implicated in the development of atherosclerosis and MI.44)45) Adiposity has also been shown to be positively associated with ischaemic and haemorrhagic stroke.44)45) Higher levels of BMI during childhood have also been shown to increase CHD risk in later life.46)47) The relationship between obesity and CVD has been found to hold in East Asian populations, and specifically in Korea.48) For example, Choi et al.49) find evidence of a positive association between obesity and weight change with respect to CHD in Young Korean adults using data from the Korean National Health Insurance Service.
In recent years, much attention has been given to the role of C-reactive protein (CRP) in the development of CVD.45)50) Whilst CRP has been identified as a biomarker for CVD related illness, it has not been clear whether CRP is causally relevant in the development of CVD, and consequently whether it would serve as an effective drug target.50)51) It has been suggested for example, that CRP could serve as a mediator in relation to LDL-C.52) Alternative assessments of the role of CRP emphasise that CRP may simply be correlated with true risk factors of CVD.53) As a consequence, while CRP is useful in predicting onset of CVD, the efficacy of interventions aimed at reducing CRP is unclear from observational studies alone.51)
Physical activity
There is growing evidence linking physical activity to CVD, and specifically CHD events.54) Sedentary individuals have been shown to have an increased risk of CVD related illness, though this could likely be driven by correlations between physical activity and alternative risk factors for CVD, such as obesity.55) However, whilst this relationship appears to hold regardless of gender, there is much debate over exercise guidelines and defining at-risk groups using arbitrary thresholds. An apparent dose-response relationship between physical activity and the reduction of CVD risk is suggested by previous research, but individuals at extreme levels of inactivity appear to be at a disproportionately high risk for CVD.56) These findings have been confirmed by several studies specific to Korea, though the extent to which physical activity is causally associated with CVD risk independent of risk factors such as obesity is uncertain.39) Using data from the South Korean health insurance system, Kim et al.57) find evidence of an inverse association between physical activity and CVD, independent of adiposity, smoking, and alcohol consumption.
Hypertension
Elevated blood pressure levels are widely accepted to be a risk factor for CVD events, based on findings from observational analyses.58)59) However, while both systolic blood pressure (SBP) and diastolic blood pressure (DBP) seem to have a continuous, independent, and positive association with CVD, it is important to highlight the difficulties in using an arbitrary threshold to classify hypertension cases.60) It has been argued that regional and demographic differences in blood pressure levels should be taken into account, in particular with respect to age where young adults exhibiting multiple risk factors may fail to reach high risk levels, owing to the strength of age as a risk factor for CVD. In East Asian populations, hypertension has been shown to be positively associated with CVD risk.61)62) Lawes et al.61) found evidence of an association between hypertension and CVD through a pooled meta-analysis of studies specific to the Asia-Pacific region. In Korea specifically, Son et al.63) observe a positive association using data from the Korean National Health Insurance system.
Type 2 diabetes
In observational analyses, diabetes and glucose intolerance have been shown to increase the risk of CVD related illness, especially in female populations.6)64)65) It is important to note, however, that type 2 diabetes is associated with many risk factors of CVD related illness, including lipids, adiposity, and hypertension as previously discussed.66) As a consequence, it can be difficult to estimate the independent contribution of type 2 diabetes to CVD whilst controlling for potential confounding variables. Insulin resistance and metabolic syndrome have also been suggested as potential mechanisms for risk factors related to CVD, though there is much debate surrounding definitions of metabolic syndrome and its efficacy as an aggregate measure relative to assessing individual risk factors.6)67) A number of studies, including the Asia Pacific Cohort Studies Collaboration (APCSC) meta-analysis project, show evidence of a strong positive association between diabetes mellitus and stroke in Asian populations, including Korea.68)69)70)
GENETICS AND METABOLOMICS OF CARDIOVASCULAR DISEASE
The emergence of large-scale genetic data has heralded a paradigm shift in epidemiology, allowing for genetic analyses contributing to our understanding of the aetiology of disease. Identifying genetic variants directly associated with a disease can illuminate potential drug targets, as well as highlight genetic predisposition to disease onset. Further, the existence of genetic variants simultaneously associated with both a disease outcome and previously identified risk factors can lend support to findings obtained from observational analyses.
In 2007, the first GWASs were published for CHD, identifying a locus on chromosome 9p21 to be robustly associated with CHD (p=5×10−8).71)72)73) GWASs of CVD have since contributed to the identification of approximately 70 susceptibility loci, as shown in Table 1. A number of these loci can be linked to risk factors previously identified, for example, approximately 20% are located near genes with known roles in lipid metabolism and 5–10% are located near genes associated with blood pressure.
Table 1
Genetic variants from genome-wide association studies associated with CVD

Af = allele frequencies; CAD = coronary artery disease; Chr = chromosome; CRP = C-reactive protein; CVD = cardiovascular disease; HDL-C = high-density lipoprotein cholesterol; IL = interleukin; LDL-C = low-density lipoprotein cholesterol; SNP = single-nucleotide polymorphism.
*Risk factor is not directly associated with lead SNP but is associated with one or more SNPs within gene region; †SNP is associated with 2 or more different lipid fractions (LDL-C, HDL-C, and triglycerides).
The discovery of genetic variants simultaneously associated with CVD outcomes and LDL-C provides evidence of a potential role of LDL-C in CVD development. However, it should be noted that several studies report variants associated with LDL-C, HDL-C, and triglycerides simultaneously (denoted lipids in Table 1). In these cases, it is not possible to clearly distinguish which lipid fraction is potentially driving the association between the variant and CVD. Hypertension and adiposity are also linked with several of the genetic variants, further supporting evidence from observational analyses. In Table 1, we include height as a risk factor as it is a component of BMI.
It is important to emphasise, however, that the extent to which genetic analyses can support observational studies is dependent upon heritability of a given risk factor. Adiposity and height are, for example, highly heritable traits as opposed to behavioural risk factors such as smoking. As such, the absence of genetic studies supporting observational analyses does not necessarily invalidate observational findings. This could for example explain the lack of genetic variants associated with CVD and physical activity. In related work using data from KCPS-II Biobank, Jung et al utilise a genetic risk score to predict CVD incidence, comparing the accuracy to a score using traditional risk factors.25) They found that combining information using both scores substantially improved predictive accuracy, suggesting there may likely be a strong genetic component to CVD in the Korean population.
Technological advances in measuring metabolites have facilitated analyses focusing specifically upon role of metabolites in disease onset and progression. As an emergent field, metabolomics focuses upon molecular changes which occur in different disease states, elucidating the underlying biological mechanisms behind disease and allowing for effective biomarkers to be identified.74)75)76) As disturbances in cardio-metabolic processes often accompany CVD, it has been possible for metabolite profiling to be applied to CVD in order to evaluate the potential role of metabolites.74)75)
Such studies have provided evidence linking a range of metabolites present in plasma levels with CVD. For example, metabolomic studies of heart failure using mouse models have highlighted the coordinated downregulation of branch chain amino acids (BCAAs) in failing hearts.75)77) Whilst these findings have been replicated in humans, metabolomic studies typically rely upon serum metabolite measures, which may suffer from issues related specificity.78) However, despite these limitations a number of potential biomarkers for CVD have been identified. These include phosphatidylserine, C16-sphingosine, N-methyl arachidonic amide, N-(2-methoxyethyl) arachidonic amide, linoleamidoglycerophosphate choline, lysoPC (C18:2), lyso-PC (C16:0), lyso-PC (C18:1), arachidonic acid, and linoleic acid.79)80)81)82)
MENDELIAN RANDOMIZATION AND CARDIOVASCULAR DISEASE
Mendelian randomization: overview
MR is a causal inference approach which corrects for bias resulting from residual confounding.83) Such bias occurs when variables serving as joint determinants of the exposure and outcome of interest are omitted from analyses, resulting in associations between the omitted variables and the outcome being erroneously attributed to the exposure. MR corrects for residual confounding by employing genetic variants as instrumental variables (IVs).83) In their seminal work, Smith and Ebrahim83) demonstrate how genetic variants can serve as IVs and illustrate how the same principles can be applied within medical research. This has led to the rapid development of formal statistical approaches, which allow for causal relationships to be identified.84)85)86)87)88)89)90)
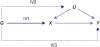
Conventional MR analyses rely upon three central assumptions for unbiased effect estimation. Firstly, genetic instruments must be strongly associated with the exposure of interest (IV1).91) Candidate instruments are typically identified using GWAS, with the extent to which an IV explains variation in an exposure of interest referred to as instrument strength. Second, genetic instruments are required to be independent of unmeasured confounders of the exposure and outcome (IV2).91) The use of genetic variants as instruments is particularly useful in this case, as the random assortment of alleles at conception restricts the range of possible associations which can occur. Third, genetic instruments must be independent of the study outcome when conditioning on the exposure of interest (IV3).92)93) Associations which violate assumption IV3 are referred to as horizontal pleiotropic pathways, inducing bias in the direction of pleiotropic association.93) The assumptions of MR are illustrated in Figure 1.
Figure 1
A directed acyclic graph illustrating the Mendelian randomization assumptions. G represents a genetic instrument, X and Y are the exposure and outcome of interest respectively, and U denotes one or more unmeasured confounders of the exposure and outcome. In the diagram, the bold arrow from G to X indicates the association between the instrument and exposure necessary to satisfy assumption IV1. The dashed arrows indicate associations which would, if non-zero, invalidate the second and third MR assumptions (IV2–3).
Mendelian randomization: current methods
Initially, MR was conceived at a point where a relative paucity of genetic data was available, owing to the scarcity and expense of GWASs. Consequently, early MR studies typically featured small sample sizes, selected genetic instruments informed by candidate gene studies, and utilised individual level data. However, the recent proliferation of GWASs and emergence of large-scale biobank projects such as the China-Kadoorie Biobank have served to motivate the wide-spread application of MR, including methods leveraging publicly available GWAS summary data.84)85)94) The availability of GWAS summary data with increasingly large sample sizes is advantageous in allowing for candidate instruments to be identified using separate non-overlapping samples. This improves the power to detect suitable instruments, as well as limiting bias due to winner's curse; occurring when a genetic variant is observed to be associated with a phenotype of interest purely by chance.
The increase in publicly available GWAS summary data has also motivated the development of myriad MR approaches which are able to furnish MR estimates without requiring access to individual level data. These methods utilise association estimates and standard errors for each genetic instrument with the exposure and outcome using separate samples, calculating a Wald ratio estimate for each genetic instrument.84)94) The Wald ratio, calculated by dividing the instrument-outcome association by the instrument-exposure association, serves as an MR causal effect estimate using a single genetic instrument. Summary level MR methods then typically evaluate the set of Wald ratios within a meta-analytic framework, through methods such as inverse-variance weighted (IVW) and MR-Egger regression.84)94)
The emergence of large-scale biobanks has also prompted the development of a range of individual level data approaches to MR. This allows researchers to incorporate more information into estimates of causal effect, such as allowing the inclusion of additional variables for adjustment and incorporating gene-by-environment interactions and non-linear models.95)96)
Mendelian randomization applications to cardiovascular disease
To assess the contributions of MR to studies focusing on cardiovascular health, we conducted a systematic review using PubMed and Web of Science. Using a comprehensive search strategy (see web appendix), we identified MR studies where CVD, coronary artery disease (CAD), CHD, heart disease, MI, or stroke served as an outcome during the period 2003–2019. All identified publications underwent a 3-step review of title, abstract, and full text based on predefined inclusion criteria. We excluded meta-analyses, review papers and studies incorporating interventions, as well as animal and laboratory studies. For each eligible study, we extracted the lead author's name, journal name, publication year, and study population. We identify a total of 126 eligible MR studies, conducted from 2003–2019, as shown in Figure 2.
Lipids
Several MR analyses have focused on the role of serum lipids, and specifically LDL-C, in the development of CVD. LDL-C has consistently been shown to be positively associated with CAD, CHD, ischemic stroke, and MI in MR analyses.97)98)99)100)101) Triglycerides have also been shown to be positively associated with CAD, CHD, and MI.97)102) It is interesting to note, however, that the observed inverse association between HDL-C and CVD has not been shown to hold in MR studies particularly in relation to CHD and MI.102)103)104)
As previously discussed, a concern when assessing the role of lipid fractions is the correlation between LDL-C, HDL-C, and triglycerides. In the context of MR, using such variants as an instrument for a single lipid fraction would likely result in bias in cases where additional lipid fractions are independently causal with respect to CVD, as this would violate the third MR assumption (IV3). Studies using variants specific to HDL-C have not found evidence of an association with CVD related illness, though this could potentially be due to a lack of enough statistical power.103)
A potential solution to this problem is the application of multivariable MR methods, which allow for multiple risk factors to be adjusted for provided they independently explain sufficient variation in each exposure.90) Several studies have implemented this research design, finding evidence of a positive association between LDL-C and CVD outcomes, whilst HDL-C does not appear to have a substantial effect.105)106)
Alcohol consumption
MR studies have also supported the observed link between alcohol consumption and CVD outcomes. Findings from the China-Kadoorie Biobank, comprised predominantly of Chinese participants, have shown a positive association between alcohol and subtypes of stroke using both MR and observational approaches.30) This observed association between stroke outcomes and alcohol consumption has also been replicated in European populations.107)
Alcohol consumption has also been linked to wider CVD outcomes in a number of MR studies including Korea specific populations.30)108)109) For example, several studies have utilised gene-by-environment MR approaches to examine the association between alcohol and CVD in East Asian populations, leveraging gender-specific differences in drinking behaviour.108)110) In an analysis using data from the Korean Genome and Epidemiology Study (KoGES), Cho et al.108) find evidence of an association between alcohol consumption and cardiovascular health. Findings from Jee et al.111) using data from KCPS-II Biobank also find evidence of an association between alcohol and hyperuricemia in males using data from KCPS-II Biobank.
Obesity and C-reactive protein
The relationship between adiposity, often measured using BMI, and CVD has also received substantial attention in the MR literature. CAD, for example, has been found to be positively associated with adiposity in a number of studies.112)113)114)115)116) Importantly, differing measures of adiposity have often been included such as waist-to-hip ratio, so as to limit the extent to which the findings are an artefact of selecting BMI as the primary measure of adiposity.113)117) Adiposity has also been found to be a risk factor for many CVD related diseases, including stroke and CHD.118)119)120) In MR analyses focusing upon the role of CRP in the development of CVD related illness, there appears to be no substantial evidence of causal association.121)122)123)124) Such findings provide support for the use of CRP as a biomarker, rather than a target for intervention.
Type 2 diabetes
Whilst many MR studies focus upon diabetes as an outcome rather than a risk factor, several studies suggest diabetes and glucose are risk factors for CVD related illness. Altered glucose levels have been found to be associated with CAD independent of adiposity and are thought to be a mechanism through which diabetes causes CAD.112)125) Diabetes has also been estimated to be positively associated with CHD and large artery stroke.126)127)
Finally, it is important to emphasise that where a binary exposure is evaluated within a summary MR framework a subtle difference in interpretation is required. Essentially, the effect estimate is not the effect of the disease itself, but rather the phenotypic effects of general liability to that disease.128)
Smoking, physical activity, and hypertension
At this point there is a relative gap in the MR literature pertaining to smoking, physical activity, and hypertension as risk factor for CVD. Studies conducted in European populations have found evidence of a positive association between SBP and CVD events, confirming findings from observational analyses.129) In a Norwegian population, smoking has been found to be a risk factor for several potential downstream risk factors of CVD, though a direct association between CVD and smoking was not examined.130) Unfortunately, there appears to be insufficient evidence from MR related studies to explore the role of physical activity in CVD.131) As with genetic analyses, MR studies are reliant upon risk factors being highly heritable, which translates to instrument strength. In the case of behavioural risk factors, it is often not possible to identify genetic variants which can explain a enough proportion of the variance of a given risk factor to provide a sufficiently precise estimate of causal effect.
PRECISION MEDICINE AND FUTURE DIRECTIONS
By assessing findings of CVD analyses using a range of study designs, it is possible to identify several relationships which are consistently present. The relationship between LDL-C and CVD remains strong across observational, genetic, and MR analyses, and across populations of differing ethnic background. However, similar agreement across studies assessing the protective effects of HDL-C is not present. There are 2 likely explanations for why this is the case. In the first instance, the observed association between HDL-C could be due to residual confounding, such that one or more risk factors are highly correlated with HDL-C but omitted from the analyses. However, as it is common practice to control for LDL-C when estimating the effects of HDL-C, identification of the omitted risk factors remains unclear. A second possibility is that the assumptions of the MR approach may be violated, leading to biased estimates. This could be the case where, for example, genetic instruments are simultaneously correlated with both HDL-C and LDL-C.
The example of LDL-C illustrates the potential to detect effective drug targets, such as the use of statins in lowering CVD related illness. Statins are medicines which specifically aim to lower serum LDL-C and have been shown to effectively reduce risks of CVD related illness. In the previous analyses, evidence for a relationship between LDL-C was found using observational, genetic, and MR methods, and the agreement across these studies of differing design highlight the therapeutic effectiveness of statins in retrospect.
A further related drug target demonstrated by genetic studies is the development of monoclonal antibodies against PCSK9.132) PCSK9 is a liver protease that targets low-density lipoprotein receptors for lysosomal degradation. Subsequent MR analyses have confirmed genetic findings, highlighting the therapeutic potential of targeting PCSK9.133) These findings have led to recommendations for low-density lipoprotein lowering with PCSK9 inhibitors to be provided for high-risk patients with LDL-C levels ≥70 mg/dL on maximally tolerated oral therapies.134) Two additional targets, CYP2C9 and VKORC1, have also been identified in GWASs, showing an association with inter-individual variability in warfarin dose.135)136) This suggests that genes in the warfarin metabolism pathway are associated with dose variance.135)136) Though these studies were performed in Japanese populations, additional studies confirmed these variants for personalized warfarin treatment in different ethnic groups.137)
Recent research has highlighted the potential for Lp(a) as a potential drug target for lowering CHD risk.138) MR analyses focusing on Lp(a) find evidence suggesting that pharmacologically lowering Lp(a) concentration by approximately 100 mg/dL can potentially reduce CHD risk by as much as 25%.138) However, the efficacy of Lp(a) as a drug target is somewhat controversial, with randomized controlled trials (RCTs) finding no substantial benefit using treatments specifically targeting Lp(a) with respect to cardiovascular events.139)140)141) The disagreement in findings between MR and RCT studies is explained in part by the limited suitability of LP(a) targeting drugs to individuals with high concentrations of LP(a). As such treatments are best suited to individuals with very high levels of Lp(a), with diminishing returns as Lp(a) concentration declines.138) This is encapsulated by the risk of CHD being estimated to be linearly proportional to the absolute difference in Lp(a) concentration across study samples.138)
Like the example of LDL-C, obesity and adiposity have been shown to be consistently associated with CVD across varying types of study. Increased BMI has been estimated to be positively associated with CVD independent of LDL-C, which is conventionally controlled for in observational analyses. Simultaneous associations between adiposity and CVD outcomes, as illustrated in Table 1, also support the existence of a causal association in conjunction with findings from MR analyses. However, it is important to highlight that substantial divergence in study findings with respect to CRP suggest drugs targeting CRP specifically will be unlikely to be effective.
The protective effects of low doses of alcohol with respect to CVD are not found to be consistent across different study designs. MR analyses performed using alcohol consumption as an exposure find evidence of a positive association between alcohol and CVD, although there are currently only a relatively small number of relevant studies. It seems possible that consumption of only small amounts of alcohol is negatively correlated with alternative risk factors, for example adiposity or type-II diabetes, which are driving the observed protective effect of alcohol through confounding bias. Further studies evaluating the causal impact of alcohol consumption would prove invaluable in elucidating the potential pathways between alcohol consumption and CVD.
There are, however, gaps in the MR literature which warrant further examination. For example, smoking, physical activity, and hypertension have all been previously identified to be risk factors for CVD related illness. However, conducting a systematic review of MR studies highlights a notable gap in the literature with respect to these risk factors. Several studies have considered these risk factors as part of wider studies related to cardiovascular health, in particular work by Burgess et al.138) and Carter et al.142) However, more work focusing on replicating MR findings is required.
Considering smoking, one potential issue is the limited number of genetic variants associated with smoking behaviour. In MR analyses, and particularly 2-sample summary analyses, the relative weakness of genetic variants as instruments for smoking behaviour results in a lack of sufficient precision to detect causal associations. However, this issue can be effectively addressed with the growing sample sizes of studies with genetic data, and the subsequent increases in precision afforded to researchers.
CONCLUSIONS
In this review we have critically examined research identifying risk factors for CVD using observational, genetic, and MR study designs. By assessing findings across multiple study designs, each with independent sources of bias, it is possible to identify consistent relationships which appear to be robust to the limitations of each research methods. This strengthens the extent to which such findings can be robust and serve as potential targets for precision medicine. We identify LDL-C, alcohol, and obesity as notable risk factors, as well as several candidate risks which warrant further attention.
 XML Download
XML Download