

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hepatic hemangioma (HH), also called infantile hepatic hemangioma (IHH), is the most common benign tumor of the liver and the third most common hepatic tumor in children [1]. IHH has historically been referred to as ‘hemangioendothelioma,’ regardless of lesion type, leading to a delay in the correct diagnosis and proper treatment of affected patients. The first classification system for IHH was developed by Mulliken in 1982 [2]. Christison-Lagay et al. [3] and Kulungowski et al. [4] refined this system and created a subtyping system for IHH with further differentiation in diagnosis that offers a more diagnosis-based treatment.
Several potentially malignant hepatic lesions may mimic HHs and require exclusion before children with IHH can be treated [5]. Serum markers such as alpha-fetoprotein (AFP) may help with the differentiation between malignant and benign diseases but could be misleading [67]
IHH shares the same growth and involution process as cutaneous hemangiomas, but most are clinically silent and remain undetected. Reasons for detection are size, location, or hemodynamic effects, but it occurs incidentally in most cases [4]. The clinical spectrum of IHH is wide, spanning from spontaneous regression with no treatment to high-volume arteriovenous shunting, secondary hypothyroidism, bleeding, abdominal compartment syndrome, and life-threatening high-output congestive heart failure with respiratory compromise. IHH is also associated with Kasabach-Merritt syndrome, presenting with hemolytic anemia, thrombocytopenia, prolonged prothrombin time, and hypofibrinogenemia [89].
Treatment options for IHH are plentiful, but no standard treatment algorithm has been determined to date. Asymptomatic lesions may undergo spontaneous regression without treatment within 1 year, whereas symptomatic lesions require medical, interventional, and/or surgical treatment modalities.
Pharmacological treatment options include steroids and interferon alfa and, more recently, propranolol. Interventional or surgical modalities include hepatic artery ligation or embolization, hepatic resection, and orthotopic liver transplantation [1]. However, due to the rarity of this disease, only a few series of patients with IHH have reported their clinical features, treatment, and outcomes. This study aimed to present the clinical experience and treatment outcomes of IHH at our center over a 5-year period.
Go to : 
MATERIALS AND METHODS
We retrospectively reviewed medical records of seven patients (four male, three female) who were treated for IHH at the University Children’s Hospital Hamburg Eppendorf, Germany, between 2010 and 2015. The following parameters were included in the analysis: age at presentation, presenting symptoms, diagnostic procedures, tumor location and size, histopathologic type, treatment modalities, and outcome.
Age at diagnosis was defined as the age at first presentation to the hospital with symptoms. Serum AFP was detected at initial presentation, and the level was interpreted as either within normal limits or elevated, considering pediatric reference values. All patients initially underwent abdominal ultrasonography (US) for diagnosis; some eventually proceeded to computed tomography and/or magnetic resonance imaging (MRI) as required after US. Percutaneous needle biopsy or open biopsy was performed when the diagnosis could not be confirmed by radiological findings alone.
Tumor location was defined as focal when a single mass was confined to one liver lobe (right or left hemilobe). The masses were defined as multifocal when multiple masses were detected throughout the liver.
In patients with clinical symptoms and a resectable tumor, primary surgical resection was performed (i.e., unilobar solitary tumor). The aim of surgery was to remove the tumor completely by anatomic resection. Liver transplantation was considered for patients with otherwise unsuccessful therapeutic approaches.
The treatment outcomes were classified as progression, in regression, complete regression, or exitus letalis. Progression was defined as increasing tumor size compared to that at diagnosis. In regression was defined as decreasing tumor size. Complete regression was defined as no tumor tissue detected at the last follow-up.
Go to :
RESULTS
Presentation
The mean age at presentation in our center was 62 days (range, –35 to 269 days; median, 3 days). One patient was diagnosed with a hepatic tumor prenatally. Three patients were diagnosed within 3 days of age. Two patients were delivered preterm. The mean follow-up duration after diagnosis was 751 days (range, 68 days to 5 years; median, 960 days) (Table 1).
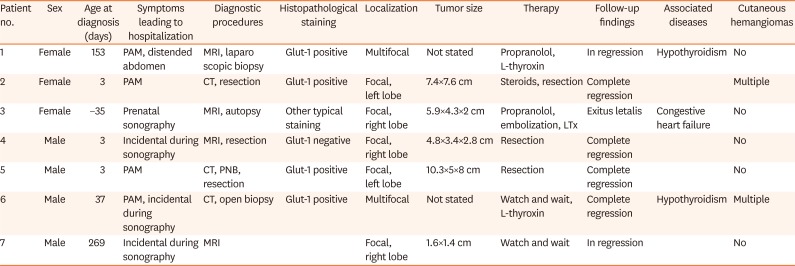
Table 1
Diagnostics, therapy, and follow-up of the patient cohort
PAM: palpable abdominal mass, MRI: magnetic resonance imaging, CT: computed tomography, PNB: percutaneous needle biopsy, LTx: liver transplantation.
![]()
The most common clinical presentation was a palpable abdominal mass (n=4) with or without a distended abdomen. Three patients had no clinical symptoms, and the hepatic lesion was diagnosed incidentally. Two patients also had hypothyroidism. The serum AFP level at presentation was within the normal range in all patients (not shown). Congestive heart failure was seen in one patient.
Diagnostics
All patients underwent abdominal US as the initial diagnostic tool. Based on the US results, three patients underwent CT scanning and four patients underwent MRI. After the radiologic examination, only two patients were diagnosed with IHH.
In the remaining cases, diagnosis could not be confirmed. Despite the radiological suspicion of IHH, biopsy and resection were performed in 5 cases (Table 1). One patient underwent US-guided percutaneous needle biopsy. Open biopsy and laparoscopic biopsy were performed for another two patients. Two patients underwent surgical tumor resection after CT or MRI and were diagnosed by analysis of the histologic specimen obtained in the surgical resection. The patient undergoing open biopsy required mechanical ventilation and catecholamine supplementation within the first postoperative day as well as two transfusions of erythrocyte concentrate during and after the intervention. The two patients undergoing needle and laparoscopic biopsy did not require further supportive treatment after the intervention.
The biopsy and resection results confirmed the diagnosis of IHH in four patients by Glut-1 staining and in one patient by typical other stains despite negative Glut-1 staining [4]. One patient was diagnosed histologically postmortem. Of all seven patients, five (81%) had a focal single tumor (three right lobe, two left lobe), while two had multifocal/bilobar disease.
Treatment
Two patients were treated with propranolol. One was dosed with up to 15 mg/kg/day and showed regression after 260 days but was lost to follow-up. The other was treated with 3 mg/kg/day but was listed for transplantation due to therapy failure and deterioration of the general condition along with congestive heart failure. The patient was too young to undergo an immediate operation. Instead, therapy with propranolol was attempted, but it did not show satisfactory outcomes. Embolization could not be performed successfully due to excessive arteriovenous intrahepatic shunts. Finally, liver transplantation was initiated, but the patient died intraoperatively due to extensive bleeding. For two asymptomatic patients, no treatment was administered, regardless of tumor extent. One patient showed no tumor after 3 years, while the other patient showed progression but then regression in further follow-up.
Three of seven patients (42.9%) underwent tumor biopsy for diagnostic reasons after non-conclusive imaging; one of underwent resection. In three of seven patients (42.9%), open surgical resection of the tumor was performed after therapeutic imaging and/or biopsy. Six of seven patients (85.7%) underwent an invasive procedure for diagnostic or therapeutic reasons.
Go to :
DISCUSSION
Over 5 years, we treated seven patients with IHH in our pediatric center for liver diseases. IHH is a rare disease; with an incidence of 5 in 1,000,000 children per year, it is difficult to perform large cohort studies [10].
Most of our patients (42%) presented with a palpable abdominal mass but no further symptoms correlating with other studies showing that 47% of patients presented with abdominal distension as the most frequent symptom. Moreover, patients are probably not diagnosed when symptoms of abdominal disease are lacking [11]. Six of our seven patients were diagnosed prenatally to 153 days of age, confirming similar findings in the literature regarding presentation within the first 6 months of life [312]. One patient was diagnosed at 269 days of age.
In our cohort, only two patients showed additional cutaneous hemangiomas (29%), an incidence that is lower than in other reports [13]. Two patients showed hypothyroidism.
In rare cases, patients show signs of Kasabach-Merritt syndrome or early congestive heart failure indicative of vast shunting volumes [1]. The incidence of life-threatening conditions in IHH is 10-20% with a high mortality rate [14]. In these cases, prompt diagnosis and intervention are necessary to prevent progression or even death.
Several other potentially fatal hepatic lesions may mimic different IHH types, including hepatoblastoma, mesenchymal hamartoma, congenital cysts (such as ciliated foregut duplication cysts), or kaposiform hemangioendotheliomas [5]. AFP is commonly used as a diagnostic marker for malignancies, but it can be misleading as its levels may be elevated in benign diseases. AFP is regularly elevated in newborns, so it cannot regularly be used to diagnose malignant diseases [715]. Serum markers must be used with caution in young children and only in combination with clinical and radiological findings.
MRI and US are the most commonly used diagnostic procedures and recommended for the imaging of abdominal masses [5]. Their use can lead to a first differentiation between malignant and benign masses. In our cohort, only one patient was diagnosed based on radiologic findings alone. This was the last patient in a series of cases; thus, it could be considered as a potential effect of learning in the process of diagnosis and treatment after repeated treatment of patients with IHH. Although not all types of IHH express Glut-1, fine needle biopsy with Glut-1 analysis will remain one of the most sensitive diagnostic tools for IHH [31617]. Although not all specimens will show typical staining patterns, the diagnosis can be made using other stains, as was observed in one case in our series.
Despite great effort made to create diagnostic algorithms and increased knowledge of the radiologic appearance, the heterogeneous appearance of IHH presents a big challenge to the physician diagnosing the disease using only these findings.
Once the diagnostic process is complete, therapeutic options stated are diverse. Efforts made by a United States Registry [18] resulted in recommendations that were recently updated in a review. In the period before IHH was identified as a benign tumor and before the era of propranolol and steroids, surgical therapy and embolization were considered the standard therapy. However, many complications can occur. Due to growing knowledge and recent findings of medical therapy options, these procedures seem obsolete and are reserved for exceptional cases, such as those with abdominal compartment syndrome or therapy failure.
Embolization is advised in rare cases of arteriovenous shunting, while medical intervention or therapy can be useful in cases of multifocal and diffuse IHH. Propranolol has been discussed several times as a therapeutic approach for infantile cutaneous hemangiomas as well as IHH [13]. Good treatment results with few side effect were reported in several case reports on children with IHH; one case even involved congestive heart failure [1920]. Experience continues to accumulate regarding the off-label use of beta-blockers to treat this particular entity. However, dosages vary in the literature and must be validated in further studies.
The large proportion of surgical interventions in our cohort is most likely an indication of uncertainty about diagnostic pathways and, in particular, a sign of problems with radiologic interpretation. Nonetheless, surgical hepatic interventions carry a high risk of side effects such as digestive tract complications and severe bleeding, which may be avoidable [21]. However, surgery and resection offer the highest certainty of diagnosis and the differentiation between malignant and benign tumors. This differentiation should be a special target for further studies [22]. Due to previous cases, continuous improvement in diagnostics and therapy has occurred in our hospital. Therefore, in the latter case, invasive procedures for diagnosis and therapy could be discarded; rather, the patient underwent only MRI and a watch and wait approach was adopted as therapy.
Despite ongoing research and increasing knowledge about IHH, the data on diagnostic procedures and therapy remain limited. Due to a lack of official guidelines, it is difficult for the physician to diagnose the disease using least invasive methods but minimize the risk of overlooking another IHH-mimicking disease. Similar difficulties arise in the disease therapy. A future objective must be to prevent overtreatment e.g. pursuing surgery instead of medical treatment. Therapy options used in former mentioned algorithms require regular discussion with all gathered short- and long-term results worldwide. No studies to date have examined medical treatment with steroids or propranolol.
To manage these questions, one large international study of IHH in progress in the US publishes strong results on a regular basis. Considering that patient management varies among nations and sometimes even within a single country, we suggest the development of a European-wide registry for IHH to address all disciplines that diagnose and treat IHH. These disciplines would include pediatric gastroenterology, pediatric surgery, pediatric oncology, and pediatric radiology. The aim of this registry is to capture the characteristics and therapeutic strategies of all patients with IHH within Europe.
Outreach involves extending the registry to other European countries and cooperating with other international studies to increase our knowledge of IHH and its differentials. For further information, please contact the author.
Go to :
 XML Download
XML Download