

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Considerations of Environmental Factors to Solve the Missing Heritability before the Introduction of Epigenetics
Definition and Mechanisms of Epigenetics
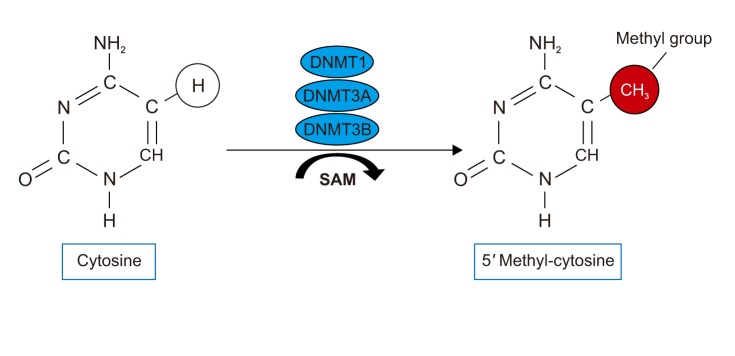 | Figure 1The DNA methylation is the covalent addition of a methyl group to a cytosine residue in a CpG dinucleotide. DNMT: DNA methyltransferase; SAM: S adenosyl methionine.
|
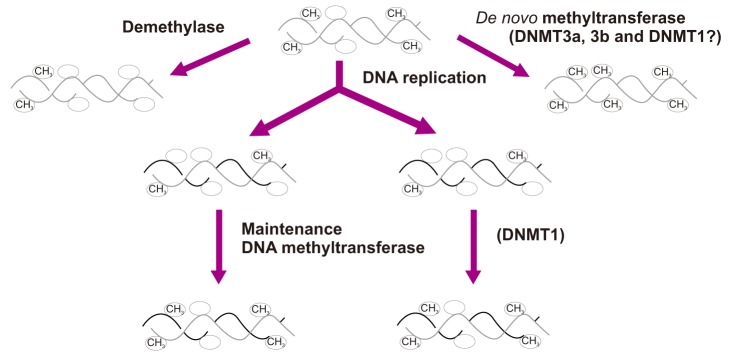 | Figure 2The DNA methyltransferase family of enzymes catalyze the transfer of a methyl group to DNA. De novo methyltransferases (DNMT3A and DNMT3B) newly methylate cytosines and express mainly in early embryo development. Maintenance methyltransferases (DNMT1) add methylation to DNA when one strand of DNA is already methylated.
|
CpG Methylation of the Airway Epithelium
Table 1
List of asthma-associated DNA methylations on candidate genes
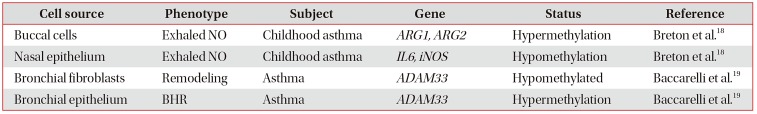
| Cell source | Phenotype | Subject | Gene | Status | Reference |
|---|---|---|---|---|---|
| Buccal cells | Exhaled NO | Childhood asthma | ARG1, ARG2 | Hypermethylation | Breton et al.18 |
| Nasal epithelium | Exhaled NO | Childhood asthma | IL6, iNOS | Hypomethylation | Breton et al.18 |
| Bronchial fibroblasts | Remodeling | Asthma | ADAM33 | Hypomethylated | Baccarelli et al.19 |
| Bronchial epithelium | BHR | Asthma | ADAM33 | Hypermethylation | Baccarelli et al.19 |
![]()
Table 2
List of asthma-associated DNA methylations on EWAS
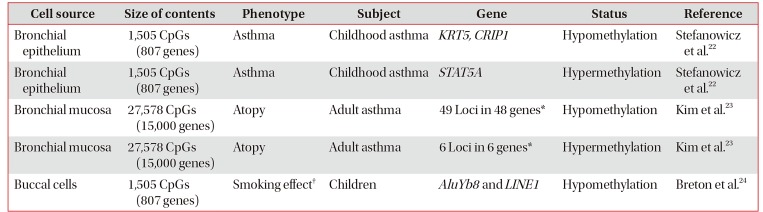
| Cell source | Size of contents | Phenotype | Subject | Gene | Status | Reference |
|---|---|---|---|---|---|---|
| Bronchial epithelium | 1,505 CpGs (807 genes) | Asthma | Childhood asthma | KRT5, CRIP1 | Hypomethylation | Stefanowicz et al.22 |
| Bronchial epithelium | 1,505 CpGs (807 genes) | Asthma | Childhood asthma | STAT5A | Hypermethylation | Stefanowicz et al.22 |
| Bronchial mucosa | 27,578 CpGs (15,000 genes) | Atopy | Adult asthma | 49 Loci in 48 genes* | Hypomethylation | Kim et al.23 |
| Bronchial mucosa | 27,578 CpGs (15,000 genes) | Atopy | Adult asthma | 6 Loci in 6 genes* | Hypermethylation | Kim et al.23 |
| Buccal cells | 1,505 CpGs (807 genes) | Smoking effect† | Children | AluYb8 and LINE1 | Hypomethylation | Breton et al.24 |
*Names of the genes and listed in Figure 3. †Prenatal cigarette smoke exposure.
EWAS: epigenome-wide association study; KRT5: keratin 5; CRIP1: cysteine rich protein 1; STAT5A: signal transducer and activator of transcription 5A; AluYb8: a short interspersed nucleotide element; LINE1: long interspersed nucleotide element.
![]()
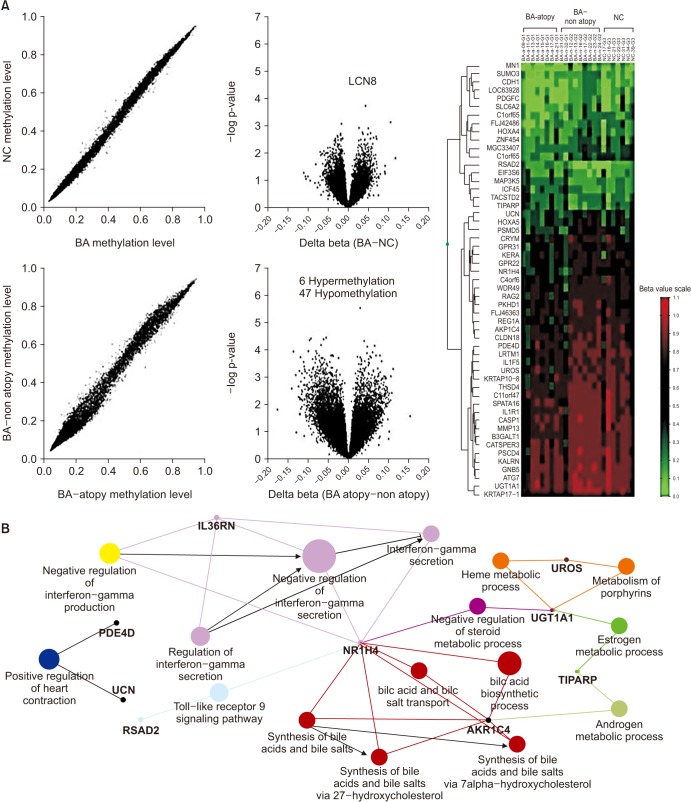
CpG Methylation of the Immune Cells
Table 3
List of asthma-associated DNA methylations of peripheral, umbilical cord blood, and bronchoalveolar lavage cells
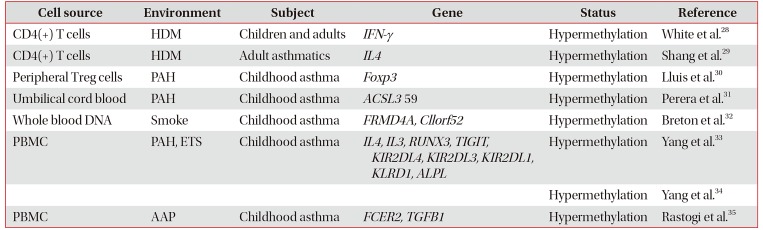
| Cell source | Environment | Subject | Gene | Status | Reference |
|---|---|---|---|---|---|
| CD4(+) T cells | HDM | Children and adults | IFN-γ | Hypermethylation | White et al.28 |
| CD4(+) T cells | HDM | Adult asthmatics | IL4 | Hypermethylation | Shang et al.29 |
| Peripheral Treg cells | PAH | Childhood asthma | Foxp3 | Hypermethylation | Lluis et al.30 |
| Umbilical cord blood | PAH | Childhood asthma | ACSL3 59 | Hypermethylation | Perera et al.31 |
| Whole blood DNA | Smoke | Childhood asthma | FRMD4A, Cllorf52 | Hypermethylation | Breton et al.32 |
| PBMC | PAH, ETS | Childhood asthma | IL4, IL3, RUNX3, TIGIT, KIR2DL4, KIR2DL3, KIR2DL1, KLRD1, ALPL | Hypermethylation | Yang et al.33 |
| Hypermethylation | Yang et al.34 | ||||
| PBMC | AAP | Childhood asthma | FCER2, TGFB1 | Hypermethylation | Rastogi et al.35 |
HDM: house dust mites; IFN-γ : interferon γ; IL4 : interleukin 4; Treg: regulatory T cell; PAH: polyaromatic hydrocarbon; ACSL3: acyl-CoA synthetase long chain family member 3; FRMD4A: FERM domain containing 4A; PBMC: peripheral blood mononuclear cell; ETS: environmental tobacco smoke; RUNX3: runt-related transcription factor 3; TIGIT: T-cell immunoreceptor with Ig and ITIM domains; KIR: killer cell immunoglobulin-like receptor; AAP: ambient air pollution.
![]()
Smoke and Air Pollutants-Related CpG Methylation and Its Mechanism
Relation of CpG Methylation with SNPs and Transcriptome
Environmental Factor-Induced Temporal Changes of CpG Methylation
Table 4
List of methylation using trajectory analysis and experimental exposure to environmental factors
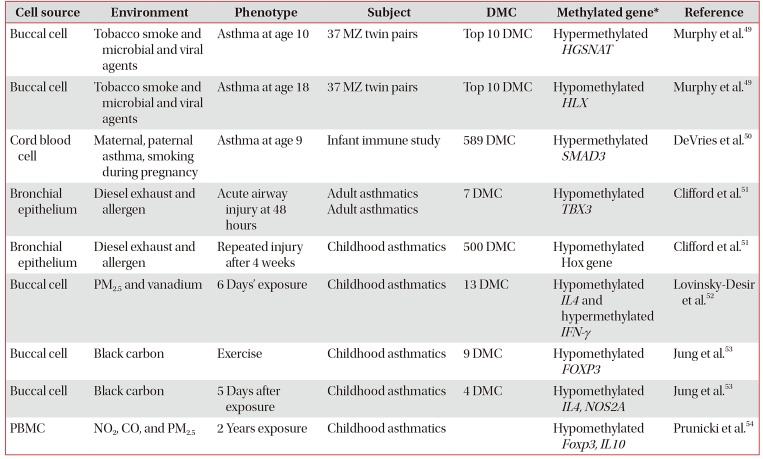
| Cell source | Environment | Phenotype | Subject | DMC | Methylated gene* | Reference |
|---|---|---|---|---|---|---|
| Buccal cell | Tobacco smoke and microbial and viral agents | Asthma at age 10 | 37 MZ twin pairs | Top 10 DMC | Hypermethylated HGSNAT | Murphy et al.49 |
| Buccal cell | Tobacco smoke and microbial and viral agents | Asthma at age 18 | 37 MZ twin pairs | Top 10 DMC | Hypomethylated HLX | Murphy et al.49 |
| Cord blood cell | Maternal, paternal asthma, smoking during pregnancy | Asthma at age 9 | Infant immune study | 589 DMC | Hypermethylated SMAD3 | DeVries et al.50 |
| Bronchial epithelium | Diesel exhaust and allergen | Acute airway injury at 48 hours | Adult asthmatics Adult asthmatics | 7 DMC | Hypomethylated TBX3 | Clifford et al.51 |
| Bronchial epithelium | Diesel exhaust and allergen | Repeated injury after 4 weeks | Childhood asthmatics | 500 DMC | Hypomethylated Hox gene | Clifford et al.51 |
| Buccal cell | PM2.5 and vanadium | 6 Days' exposure | Childhood asthmatics | 13 DMC | Hypomethylated IL4 and hypermethylated IFN-γ | Lovinsky-Desir et al.52 |
| Buccal cell | Black carbon | Exercise | Childhood asthmatics | 9 DMC | Hypomethylated FOXP3 | Jung et al.53 |
| Buccal cell | Black carbon | 5 Days after exposure | Childhood asthmatics | 4 DMC | Hypomethylated IL4, NOS2A | Jung et al.53 |
| PBMC | NO2, CO, and PM2.5 | 2 Years exposure | Childhood asthmatics | Hypomethylated Foxp3, IL10 | Prunicki et al.54 |
*Representative genes are presented: HGSNAT (heparan alpha glucosamine N-acetyltransferase), HLX(homeobox protein HB24), SMAD3 (SMAD family member 3), TBX3 (T-Box transcription factor TBX3), Hox (homeobox), IL4 (interleukin 4), IFN-γ (interferon γ), FOXP3 (forkhead box p3), NOS2A (nitric oxide synthase 2).
MZ: monozygotic; DMC: differentially-methylated CpG site; PM2.5: particulate matters especially smaller than 2.5 µM; PBMC: peripheral blood mononuclear cell.
![]()
Global Changes in CpG Methylation of Nasal Polyps from Subjects with NERD
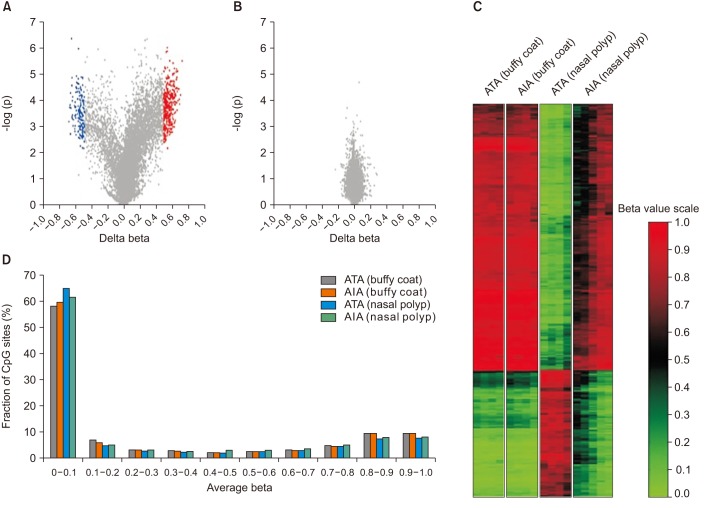 | Figure 4Summary of DNA methylation data. (A, B) Volcano plot of differential methylation level between aspirin-induced asthma (AIA) and aspirin-tolerant asthma (ATA) in nasal polyp tissues (A) and buffy coat samples (B). Red dots: delta-beta≥0.5 and p≤0.01, blue dots: delta beta≤−0.5 and p≤0.01, grey dots: −0.5≤Deltabeta≤0.5 and p>0.01. Delta-Beta: difference of DNA methylation level (subtracting the DNA methylation level of ATA from AIA). −log(p): log-transformed t-test p-values. (C) Distribution of the DNA methylation level of AIA and ATA in buffy coat and nasal polyp. Average beta: DNA methylation level (0 to 1). (D) Heatmap of 490 differentially methylated CpGs between AIA and ATA in buffy coat and nasal polyp.
|
Table 5
Number of DMG in arachidonic acid pathways in NERD compared to those in ATA
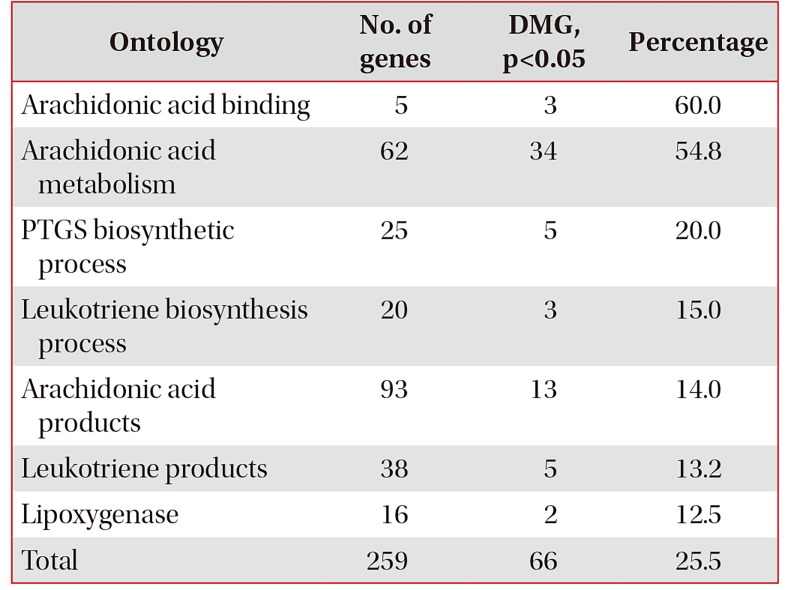
Ontology classification of the 36,127 genes in the AmiGo2 (http://amigo.geneontology.org/amigo/search/ontology) includes 259 genes in arachidonate pathways. The number of 437 DMG constitutes 1.20% of the total 36,127 genes and 66 DMG constitutes 25.5% of 259 genes in the arachidonic acid pathways.
DMG: differently methylated genes; NERD: nonsteroidal anti-inflammatory exacerbated respiratory disease; ATA: aspirin-tolerant asthma; PRGS: prostaglandin-endoperoxide synthase.
![]()
 XML Download
XML Download