

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Creutzfeldt-Jakob disease (CJD) is a rare degenerative disease with a low prevalence rate of approximately 0.5–1 per million.1 However, the survival period is generally less than a year, demonstrating its fatalness. An altered isoform of the normal cell-surface prion glycoprotein (PrPc), which is found in all mammals, is believed to be the cause of the disease.
CJD can be divided into sporadic CJD (sCJD) and variant CJD (vCJD); sCJD accounts for approximately 90% of CJD worldwide, and its peak incidence was observed in the 70s. The typical symptoms of sCJD include a rapidly progressive cognitive decline, ataxia, and myoclonus, which finally progresses to an akinetic mute state. Other early symptoms, however, have also been reported.2
Unlike sCJD, the mean age of vCJD is 29,3 and 60% to 65% of patients have psychiatric symptoms, such as depression, apathy, anxiety, social withdrawal, and agitation as initial presentation.456 In addition, patients with vCJD can develop sensory abnormalities without having any other abnormal findings. Fortunately, vCJD has not been reported in South Korea yet. In this article, we report a case of probable sCJD with extrapyramidal symptoms as the main symptom, rather than a cognitive decline.
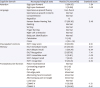
A 71-year-old male patient visited the clinic with a history of memory loss and a hollow feeling that had begun 2 months earlier. He was able to perform his daily life activities. He did not show any abnormalities during a neurological examination. His score in the Korean version of the Mini-Mental State Exam (K-MMSE) was 25, which was in the 23rd percentile, considering his age and level of education. The Seoul Neuropsychological Screening Battery (SNSB) showed that his executive function was decreased to the 1.33 percentile. Other domains were within the normal range (Table 1). He was diagnosed with non-amnesic mild cognitive impairment.
Two months later, he revisited the clinic with newly developed symptoms. His family members reported that his movement was slower than earlier. They also complained that he had visual hallucinations and that his memory impairment was worsening. His symptoms deteriorated to the point that he had difficulties in performing daily life activities. During the neurological examination, rigidity and bradykinesia were observed almost symmetrically on both sides. Resting tremor was mildly developed in his left upper extremity. His facial expression was remarkably reduced, presenting a masked face. In addition, he demonstrated postural instability, which mildly affected his daily life. He was diagnosed with Parkinsonism based on the above results. His Unified Parkinson's Disease Rating Scale (UPDRS) part III score was 26, and H&Y stage was 2.5. Total score of bradykinesia was 9 points and that of rigidity was 7 on the UPDRS part III score. He was not taking any prescribed drugs that could cause these symptoms. Unlike patients with typical idiopathic Parkinson's disease, he showed a very rapid progression of movement symptoms. Visual hallucinations during the early stage of the disease was another atypical finding.
After admission, no abnormalities were noted in the complete blood count, electrolyte level, autoantibodies, or urine following a laboratory test. Antibody screening for autoimmune encephalitis also showed negative results. Diffusion-weighted imaging and fluid-attenuated inversion recovery (FLAIR) detected abnormally high signal intensities in both the parietal and temporal lobes (Fig. 1). Despite these findings, no definite abnormalities were observed in his basal ganglia. During electroencephalography (EEG), theta and delta waves were observed in the frontal areas, but periodic discharges were not observed (Fig. 2). The results of an 18F-FP-CIT positron emission tomography (PET) imaging was normal (Fig. 1). The K-MMSE was done again, and his score was 22, showing a mild decrease from his previous score. No red or white blood cells were seen in the cerebrospinal fluid, but the fluid was positive for 14-3-3 protein. Unfortunately, the real-time quaking induced conversion test and pathological confirmation were not available. The patient's symptoms gradually worsened, and he reached a state of complete akinetic mutism after approximately 3 months.
Although a pathological diagnosis was not made, the patient's clinical symptoms and test results were consistent with the diagnostic criteria of probable CJD with three clinical features (i.e., extrapyramidal signs, visual hallucination, and progression to akinetic mutism) and two positive laboratory tests (positive 14-3-3 assay with a disease duration of less than 2 years, and high signal intensities in the parietal and temporal areas).
CJD needs to be distinguished in cases of rapid progressive dementia, and early diagnosis is essential because of its potential fatalness. The gold standard for CJD diagnosis is pathological confirmation, which is difficult to achieve when the patient is alive. The diagnostic criteria for probable CJD focus on typical symptoms, such as myoclonus and akinetic mutism. These symptoms are difficult to diagnose early, because they are often absent during the early stages of the disease.7
According to a study published in 2006, the most common initial symptom among the 114 patients diagnosed with CJD was cognitive dysfunction, including memory impairment. Nine percent of the patients complained of motor dysfunction, and approximately 25% of them complained of extrapyramidal symptoms similar to that observed in the patient in our study. In addition, the patients with CJD exhibited several neurological symptoms, including sensory symptoms and diplopia, during the early stages of the disease.2 Because of the diversity of the initial symptoms, diagnosis is difficult. Several cases of CJD with similar movement symptoms have been reported.8 Moreover, other movement symptoms such as chorea have been reported in patients with CJD.9
These motor symptoms occur because of the malfunction of several structures, including the striato-pallidal complex, mesencephalon, and thalamus. In contrast to previous reports of presynaptic dopaminergic deficits observed during FP-CIT single-photon emission computed tomography (SPECT) in CJD patients with Parkinsonism,10 we did not observe dopaminergic deficits in our patient during FP-CIT PET. This suggests that postsynaptic alterations may also contribute to the expression of movement symptoms during CJD. In addition, unlike our patient, the initial symptom of the patient in the previous report was not Parkinsonism. She demonstrated mild weakness in her left upper extremity with dysarthria at the beginning, being aggravated to clonic partial seizure combined with Parkinsonism and alteration in mental status. We carefully suggest that the invasion of the dopaminergic pathway of CJD may cause the presynaptic dysfunction afterwards, beginning with postsynaptic alteration.
As mentioned above, early diagnosis of CJD is quite challenging because of the atypical characteristics of the initial symptoms. Therefore, studies have been done using tests with diagnostic value. Real-time quaking induced conversion (RT-QulC) tests were recently introduced and have been included in the new diagnostic criteria because of their high sensitivity and specificity.1112 However, these tests are not yet widely used in South Korea and were not available in this case. Moreover, pathological confirmation was not done with the patient. Therefore, the possibility of misdiagnosis cannot be completely excluded. Despite these limitations, the clinical features and other laboratory findings including 14-3-3 protein and magnetic resonance diffusion-weighted imaging satisfied the diagnostic criteria of probable sCJD in this patient.
This case is meaningful in suggesting that diagnosis of CJD needs to be considered if the motor symptoms are rapidly exacerbated without clear cognitive impairment. Furthermore, it is noteworthy to mention the possibilities of postsynaptic alterations in movement symptoms of CJD through this case.


 XML Download
XML Download