

 PDF
PDF Citation
Citation Print
Print



Abbreviations
ADCC
antibody-dependent cellular cytotoxicity
CDC
complement-dependent cytotoxicity
CDR
complementarity determining regions
CSC
cancer stem cell
CTX
cyclophosphamide
EGFR
epithelial growth factor receptor
Fc
fragment crystallizable
HCC
hepatocellular carcinoma
HER2
human epidermal growth factor receptor 2
HMGB1
high-mobility group box 1
i.p.
intraperitoneally
i.v.
intravenously
ICB
immune checkpoint blockade
L
length
MTS
CellTiter 96® AQueous One Solution Cell Proliferation Assay
NSCLC
non-small-cell lung carcinoma
PBST
PBS containing 0.1% Tween 20
POSTECH
Pohang University of Science and Technology
TME
tumor microenvironment
W
width
INTRODUCTION
mAbs have been increasingly used as antitumor agents in clinics. Many antitumor mAbs directly target antigens expressed on tumor cells. The major targets are growth factor receptors that are overexpressed in tumors. For example, cetuximab (Erbitux®) targets epithelial growth factor receptor (EGFR) to treat colorectal cancer. Additionally, trastuzumab (Herceptin®) targets human epidermal growth factor receptor 2 (HER2) to treat breast cancer. Lineage-specific surface antigens are also targets for antitumor mAbs. Rituximab (Rituxan®) is used for treating Non-Hodgkin lymphoma by targeting CD20, which is expressed on B cells (1). These results suggest that targeting tumor-specific antigens by mAbs is a promising method of anticancer therapy. However, more targets need to be identified to generate multi-targeting strategies to prevent treatment resistance and improve patient outcome.
Other types of mAbs involve different antitumor mechanisms. They do not directly target tumor cells, but regulate neighboring cells in the tumor microenvironment (TME). For example, a specific mAb against VEGF-A, bevacizumab (Avastin®), is used to treat several types of cancer by inhibiting tumor angiogenesis. Several mAbs are designed to unleash CD8+ T cells, which often have dysregulated cellular functions due to T cell exhaustion in the TME. These mAbs target co-inhibitory molecules on T cells to operate as an immune checkpoint, reducing T cell activity under chronic viral infections and tumors. Thus, blocking immune checkpoint molecules has been pursued as a method for treating cancer with 20%–50% overall response rates (2). Ipilimumab, a mAb against CTLA-4, is the first immune checkpoint blockade (ICB) mAb to be clinically validated for metastatic melanoma and advanced non-small-cell lung carcinoma (NSCLC) (3). Nivolumab and Atezolizumab, specific to PD-1 and PD-L1, respectively, have been clinically proven for treating a substantial number of tumor types, including metastatic melanoma and NSCLC with robust responses and minimal side effects. However, its use is limited to PD-L1-positive cancer patients, where some patients reported acquired resistance by upregulating other co-inhibitory molecules (45).
New antitumor therapy approaches target cancer stem cells (CSCs) (678), because 90% of the cancer patient death is caused by CSC-dependent metastasis (910). CSCs are a subpopulation of neoplastic stem-like cells that induce malignant growth to form tumor masses that can be resistant to chemotherapy and radiotherapy. Based on these properties, CSCs play a major role in tumor-initiation, malignancy, resistance to therapy, and recurrence in both primary tumors and distant metastases, even after achieving complete remission (101112). Thus, numerous studies have addressed CSC specific markers and the development of targeted therapeutics against these antigens. Selective elimination of CSCs could lead to spontaneous regression of established tumors and prevent recurrence and metastasis (1314).
Our previous study revealed that KIAA1114, a full-length translational product of the trophinin gene, is a distinct and stable marker for CSCs in hepatocellular carcinoma (HCC) (15). Compared to other well-established markers of liver CSCs, expression level of KIAA1114 positively correlate to tumorigenic ability, independent of the HCC subtype. Furthermore, we detected expression of KIAA1114 in various human and murine cancer cell lines, suggesting that Kiatomab, a mAb specific to KIAA1114, targets CSC to treat cancer. In this study, we show that Kiatomab treatment presented antitumor responses in metastatic and subcutaneous murine tumor models. Moreover, the combined treatment with Kiatomab and cyclophosphamide (CTX) further improved antitumor effects. These results present therapeutic potential of Kiatomab as a novel mAb for anticancer therapy.
MATERIALS AND METHODS
Mice
Female BALB/c and C57BL/6 mice were purchased from Charles River Breeding Laboratories (Kanagawa, Japan). NOD/SCID strain was obtained from The Jackson Laboratory (Bar Harbor, ME, USA). All animals housed under specific pathogen-free conditions in an approved animal facility at Pohang University of Science and Technology (POSTECH) Biotech Center. All mouse experiments were performed in accordance with the National Institutes of Health guidelines, and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) guidelines of POSTECH (POSTECH-2016-0079-R2).
Cell lines and the generation of Kiatomab and its isotype variant
CT26, B16-F10, Tramp-C1, Renca, and Hepa-1c1c7 mouse cancer cell lines were purchased from American Type Culture Collection (Manassas, VA, USA). CT26-Her2/neu and MIH-2 mouse cancer cell lines were generously provided by Dr. Young Chul Sung in POSTECH (Pohang, Korea). Kiatomab was generated as described in our previous study (15), and its isotype control IgG2b was purchased (clone MPC-11; Bio X cell, West Lebanon, NH, USA). For isotype switch variant generation, a heavy chain fusion gene encoding heavy chain variable region of Kiatomab and murine IgG2a constant region and a light chain fusion gene encoding light chain variable region of Kiatomab and murine κ chain constant region were inserted into a single pAD11 vector having a dual promoter. The vector was transfected into CHO/DHFR−/− cells, and the stable cell line producing IgG2a variant of Kiatomab was generated as previously described (16). Kiatomab and its isotype switch variant were purified with HiTrap Protein G HP column (GE Healthcare, Piscataway, NJ, USA) using the AKTA purifier system. For in vitro effect of Kiatomab, the viability of tumor cells was tested by CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega, Madison, WI, USA).
Epitope determination and sequencing variable regions in Kiatomab
Peptide synthesis of potential linear epitopes was performed by Peptron, Inc. (Daejeon, Korea). To perform direct ELISA, 96-well immunoplate (Nunc Cell Culture, Waltham, MA, USA) was firstly coated with 1 µg of each candidate peptide diluted in 0.1M sodium bicarbonate buffer (pH 9.6) overnight at 4°C. Wells were washed 4 times with PBS containing 0.1% Tween 20 (PBST) and incubated with PBST containing 5% nonfat dry milk (blocking buffer) at room temperature for 1 h. After washing, various doses of Kiatomab diluted in blocking buffer were added to corresponding wells and maintained at room temperature for 5 h. After additional washing, wells were incubated with blocking buffer containing HRP-conjugated anti-mouse IgG2b (Bethyl, Montgomery, TX, USA) at room temperature for 1 h. After washing five times, TMB One Component HRP Microwell Substrate (SurModics, Eden Prairie, MN, USA) solution was added to each well and incubated for 15 min. The absorbance at 450 nm was measured with VersaMax ELISA Microplate Reader (Molecular Devices, San Jose, CA, USA).
For sequencing its variable regions, extraction of mRNA from hybridoma cells and subsequent synthesis of cDNA was performed using Total RNA Extraction Kit (Intron Biotechnology, Seongnam, Korea) and QuantiTect Reverse Transcription Kit (Qiagen, Germantown, MD, USA), respectively. cDNA was subjected to PCR amplification using variable region-specific primers. Amplified products were subjected to gel electrophoresis, isolated using Mega-spin Agarose Gel DNA Extraction Kit (Intron Biotechnology, Seongnam, Korea), and inserted into pGEM-T Easy Vector (Promega) according to the manufacturer's manual. Analysis of sequences was performed by Cosmo Genetech (Seoul, Korea) using T7 and SP6 primers.
Tumor models and treatments
For pulmonary metastasis model, 2×105 CT26 or CT26-Her2/neu cells resuspended in 100 µl PBS were intravenously (i.v.) injected into BALB/c or NOD/SCID mice. In the case of B16F10 model, the same number of cells were i.v. injected into C57BL6 mice. One day later, mice were intraperitoneally (i.p.) treated with different doses of Kiatomab, mouse IgG2b isotype control antibody, or F(ab′)2 fragment of Kiatomab in a dose-dependent manner. F(ab′)2 fragment was generated by using F(ab′)2 Preparation kit (Pierce Protein Biology, Waltham, MA, USA). For combination treatment with CTX (Baxter Healthcare S.A., Opfikon, Switzerland), 30 mg/kg CTX was administered i.p. at day 4. The lungs were harvested and the number of metastatic tumor nodules was determined 2 wk after tumor inoculation.
In a subcutaneous solid tumor model, 5×105 CT26 cells resuspended in 100 µl PBS were subcutaneously injected into BALB/c mice. The tumor-bearing mice were i.p. injected with indicated doses of Kiatomab or its isotype switched variant day 4 and 11 after tumor inoculation, and 30 mg/kg CTX was treated on the same day. Tumor masses were measured every 3 days by assessing length (L) and width (W) using a digital caliper. Tumor volume was calculated using the formula (V=1/2*L*W2). All experiments were terminated when tumors reached a mean volume of 1,000 mm3.
Flow cytometry
All tumor cells were incubated with fragment crystallizable (Fc)-blocker (eBioscience, San Diego, CA, USA) to prevent non-specific antibody binding and then stained with Kiatomab or mouse IgG2b isotype antibody (MPC-11, Biolegend, San Diego, CA, USA) and subsequently stained with anti-mouse IgG2b (RMG2b-1, Biolegend).All samples were acquired with LSR Fortessa (BD Biosciences, San Jose, CA, USA) and analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
RESULTS AND DISCUSSION
Identification and characterization of the linear epitope and variable regions of Kiatomab
In our previous study, we found that Kiatomab was able to recognize linear KIAA1114 epitopes that are commonly expressed in humans and mice (15). Therefore, overlapping amino acid sequences were identified, and corresponding peptides were synthesized for epitope mapping. In detail, the N-terminus (amino acids 1–366) sequence of human KIAA1114 was aligned with that of mouse KIAA1114, and the matching sequences were identified. Among them, possible linear epitope sequences in Fig. 1A were determined by Emini surface accessibility prediction and BepiPred programs (17). Peptides of regions identified by both algorithms were synthesized. Direct ELISA assays using various candidate peptides showed that residues 200 to 214 were specifically recognized by Kiatomab, and that its binding was concentration-dependent (Fig. 1B and C).
Figure 1
Characterization of Kiatomab and KIAA1114 expression in mouse cancer cells. (A) Amino acid sequences of the extracellular domain of human KIAA1114 and corresponding mouse KIAA1114 sequences were aligned. Among their overlapping sequences, possible linear epitope regions were determined by surface accessibility prediction (dotted lines) and Bepipred linear epitope prediction (solid lines) programs. Subsegments simultaneously identified by two prediction tools were synthesized as peptides (boxes) and subjected to direct ELISA using Kiatomab. (B) The binding of Kiatomab at 3µg/ml to various peptides and (C) dose-dependent Kiatomab binding are shown. (D) The variable regions of Kiatomab were clarified by RT-PCR with mRNA from the hybridoma, and CDR was determined through the Kabat sequence database. (E) KIAA1114 expression in various murine cancer cell lines was determined by Kiatomab. Representative data are shown from two or three independent experiments.

In addition, the variable region sequences of Kiatomab were determined by reverse transcription PCR using mRNA extracted from the hybridoma using variable region-specific primers shown in Fig. 1D. Complementarity determining regions (CDR) were identified based on Kabat sequence definition (18). Sequence analysis further revealed that the variable region of the heavy chain of Kiatomab showed 98% identity in both nucleotide and amino acid sequences with a germline gene, IGHV5-6*01 (IMGT annotation). In the variable region of the light chain, Kiatomab displayed 99% nucleotide and 96% amino acid identity with a germline sequence, IGKV8-30*01.
Kiatomab inhibits metastasis of KIAA1114-overexpressing cancer cells through the host immune system
We measured KIAA1114 expression after Kiatomab treatment in various murine cancer cell lines. Among the seven murine cell lines evaluated in Fig. 1E, CT26 and CT26 HER2/neu murine colon cancer cell lines were selected as representative targets based on their high levels of KIAA1114 expression. First, we determined whether in vitro treatment with Kiatomab could inhibit proliferation of cancer cell lines. Using various concentrations of Kiatomab, no direct effects on proliferation of cancer cells was observed, suggesting that Kiatomab does not directly transduce growth-inhibiting, or apoptosis-inducing signals in target cancer cells (Fig. 2A). To further assess the therapeutic potential of Kiatomab, we tested whether in vivo Kiatomab treatment eliminated KIAA1114-expressing cells in syngeneic transplant models. In experimental metastasis models, 2×105 cells of each cell line were by i.v. injection into BALB/c mice. Treatments were performed by i.p. injection 24 h after tumor inoculation. Fourteen days after tumor inoculation, Kiatomab treatment suppressed the formation of metastatic nodules in a dose-dependent manner (Fig. 2B). Kiatomab treatment at a dose of 1 µg resulted in a 2-fold decrease in the number of metastatic nodules. A single 10 µg dose exerted strong inhibitory effects on lung metastasis, as shown by more than 4- and 14-fold reductions in the number of pulmonary nodules in CT26-HER2/neu and CT26 tumor-bearing mice, respectively. Anti-metastatic effects of Kiatomab were also identified when B16F10 mouse melanoma cells were infused by i.v. injection in C57BL/6 mice (Fig. 2C).
Figure 2
Immune-mediated suppression of tumor growth by Kiatomab in pulmonary metastasis models. (A) Different doses of Kiatomab or control Ig were co-cultured with tumor cells for 72 h. The viability of cells was determined by MTS assay. (B-E) 2×105 tumor cells were i.v. injected into (B) BALB/c, (C) C57BL/6, and (D, E) NOD/SCID mice. One day later, the indicated doses of Kiatomab (10 µg for NOD/SCID mice), its F(ab′)2 fragment, or control Ig were i.p. injected into tumor-bearing mice. On day 14 after tumor injection, lungs were extracted and the number of metastatic nodules on the lung surface was counted (n=7). (E) Tumor-bearing mice were treated with 10 µg of Kiatomab and control Ig at the indicated time points.
Data are representative of three independent experiments and shown as mean±SEM.
*,#p<0.05; **,##p<0.01; ***p<0.001 versus corresponding control Ig-treated groups by Mann-Whitney U test.

We observed no anti-metastatic effects in mice treated with 7.6 µg F(ab′)2 fragment of mAb, which was equivalent to 10 µg Kiatomab in terms of molar concentration (Fig. 2B). In addition, Kiatomab treatment in NOD/SCID immune-deficient mice failed to induce anti-metastatic effects, in contrast to immunocompetent BALB/c mice (Fig. 2D). These results suggested that primary anti-metastatic mechanisms of Kiatomab are likely to involve antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Next, we assessed optimal treatment time-point for Kiatomab infusion in CT26 and CT26 HER2/neu pulmonary metastasis models. Mice treated with Kiatomab after 24 h showed a significant reduction in the number of pulmonary nodules, while mice treated after 72 h showed no effects (Fig. 2E). These results suggest that Kiatomab exerts antitumor activity during the early phases of metastasis.
Combination with Kiatomab and CTX enhances the antitumor efficacy in both metastasis and solid cancer models
Since CTX treatment is known to enhance ADCC (1920), CTX was applied in combination with Kiatomab treatment in metastatic models to induce synergistic antitumor effects (Fig. 3A). Similar to the previous results, Kiatomab treatment showed antitumor activity when applied at early time points after tumor cell inoculation (Fig. 3B). A 30 mg/kg dose of CTX alone at 4 days after tumor cell inoculation also displayed mild antitumor effects (Fig. 3B). The combination of Kiatomab (day 0) and CTX (day 4) almost completely suppressed pulmonary nodule growth, leading to enhanced survival compared to mice treated with a single agent (Fig. 3B and C). Interestingly, synergistic anticancer effect of Kiatomab was also observed when applied on day 3 together with CTX, suggesting that CTX treatment reinforces Kiatomab activity in later stages of metastasis. In another lung metastasis model with B16F10 melanoma cells, combination treatment generated synergistic antitumor effects in a similar manner (Fig. 3D). In contrast to the CT26-HER2/neu model, single treatments with Kiatomab (day 3) or CTX (day 4) showed no antitumor effects in the B16F10 model. However, combined treatments significantly reduced the number of pulmonary nodules.
Figure 3
The combination of Kiatomab with CTX improves antitumor activity in metastatic tumor models. (A-C) 2×105 CT26-HER2/neu or (D) B16F10 tumor cells were i.v. injected into BALB/c or C57BL/6 mice. Tumor-bearing mice were i.p. treated with 10 µg Kiatomab at day 0 or day 3, 30 mg/kg CTX at day 4, or both agents at the designated time points. Control group was injected with control Ig or PBS for CTX. Mice were either sacrificed (B, D) at day 14 for metastatic nodule counting (n=7) or (C) maintained for 10 wk for survival monitoring (n=10).
Data are representative of three independent experiments and shown as mean±SEM.
*p<0.05; **p<0.01; ***p<0.001 by Mann-Whitney U test or log-rank test.

Next, we tested whether Kiatomab treatment could induce antitumor effects in solid tumor models. In contrast to tumor suppression observed in the metastasis model, the administration of the same dose of Kiatomab did not induce antitumor effects in CT26 solid tumors (Fig. 4A). However, CTX treatment showed significant antitumor activity in solid tumors, and the combination of Kiatomab with CTX further enhanced efficacy (Fig. 4A). To improve therapeutic efficacy of Kiatomab, an IgG2a isotype switch variant was generated by linking the gene encoding the variable region of the heavy chain of Kiatomab to cDNA encoding mouse IgG2a constant region without modifying the light chain sequence. A number of studies have shown that IgG2a variants can exert stronger antitumor effects than IgG2b variants derived from the same mAb. This is due to the ability of IgG2a to bind to the activating Fc receptor, FcγRI, with higher affinity; and to inhibitory receptor, FcγRIIB, with lower affinity compared to IgG2b (2122). Also, IgG2a isotype has a longer in vivo half-life than IgG2b isotype (23). As expected, isotype switching of Kiatomab to IgG2a markedly enhanced antitumor effects (Fig. 4B). Injection of two 10 µg doses of the IgG2a variant induced a similar magnitude of tumor growth inhibition as two 30 µg doses of Kiatomab. Moreover, mice treated with the IgG2a mAb at equivalent Kiatomab doses showed more than 2.5-fold reduction in tumor volume compared to Kiatomab-injected mice at the end of the monitoring period (day 18).
Figure 4
CTX combination and isotype switching augment the antitumor activity of Kiatomab in a solid tumor model; 5×105 CT26 cells were subcutaneously injected into BALB/c mice. (A) On day 4 and 11 after tumor inoculation, mice were i.p. treated with 30 µg Kiatomab, 30 mg/kg CTX, or a combination of 2 agents. Tumor growth was monitored until the mean of PBS-treated group reached 1,000 mm3 (day 18) (n=7). (B) On day 4 and 11, mice were injected with 30 µg of Kiatomab, 10 or 30 µg of IgG2a isotype switch variant. Mice were maintained up to 18 days for tumor growth measurement (n=7).
Data are representative of 3 independent experiments and shown as mean±SEM.
*p<0.05; **p<0.01 by 2-way ANOVA test.
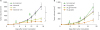
In this study, we demonstrated that Kiatomab, a novel mAb specific to CSC antigen KIAA1114, elicited antitumor responses for both metastatic and solid tumors. Antitumor activity of Kiatomab was shown in mouse models of colorectal cancer and melanoma in BALB/c and C57BL/6 mice, respectively. The elucidation of exact mode of action of Kiatomab requires further study. However, specific binding to KIAA1114 antigen on tumor cells, Fc-mediated cytotoxicity, and cell-mediated cytotoxicity likely mediate antitumor activity. Since many immune cell types are involved in the Fc-mediated antitumor responses, specific targeting of immune cells remains to be examined for better understanding of the action of Kiatomab. Co-treatment with CTX improved antitumor activity of Kiatomab, even in a solid tumor model. Previous studies proposed that metronomic CTX therapy could increase the permeability of endothelial cells of tumor neovasculature (2425). Therefore, we hypothesize that low-dose CTX treatment improved penetration of anti-KIAA1114 mAb into target tissues, thereby increasing the local concentration of Kiatomab in tumors. Another possible explanation for synergistic effects is sensitization of tumor cells to CTX mediated by anti-KIAA1114 mAb treatment. A prior study of human colorectal cancer identified a gene that showed a strong positive correlation with trophinin, high-mobility group box 1 (HMGB1), which is known to confer resistance to chemotherapy in diverse cancer models (262728). Based on the parallel pattern of expression between KIAA1114 and trophinin (29), HMGB1-expressing drug-resistant tumor cells may also express KIAA1114. Thus, Kiatomab treatment may enhance the therapeutic performance of chemotherapy by selectively eliminating these resistant cells. Combined treatment with Kiatomab and other immunotherapy regimes, like ICB, will be an interesting topic for future study. Furthermore, the therapeutic potential of Kiatomab needs to be addressed in fully-established or spontaneous tumor models.
 XML Download
XML Download