

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor,
High-molecular-weight kininogen (HK) circulates bound to prekallikrein(PK), which can bind to negatively charged phospholipids.It is a non-enzymatic cofactor for kallikrein binding to negativelycharged phospholipids. Kallikrein activates factor XII, which then activates factor XI, leading to activation of the intrinsic coagulationpathway [1].
Deficiencies in HK and PK can cause isolated activated partialthromboplastin time (aPTT) prolongation with normal levels of intrinsic coagulation factors (VIII, IX, XI, and XII). In PK deficiency,pre-incubation of patient plasma with an aPTT reagent before aPTT assay shortens the aPTT, whereas in HK deficiency, such pre-incubation does not correct aPTT prolongation [2]. Severalcases of HK deficiency have been reported in the Japanese population [3]. Herein we report the first Korean case of congenitalHK deficiency that resulted in a prolonged aPTT without a change in the pre-incubation aPTT assay. HK deficiency was confirmed by plasma HK levels and identification of two pathogenicvariants, one of which was novel (c.488delG). This study was approved by the institutional review board (IRB) of Seoul National University Hospital (IRB 1911-116-1080). Informed consent for performing genetic testing along with additional coagulationassays was obtained from the patient.
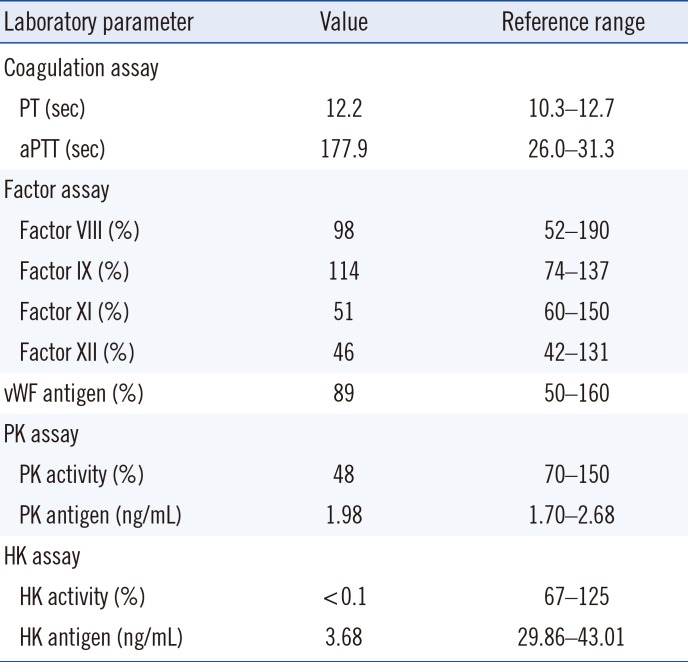
A 37-year-old man visited the CHA Bundang Medical Center, Seongnam, Korea, for lipoma surgery. He underwent preoperativeassessment, including coagulation assays, during which aPTT prolongation was found. He had suffered from peptic ulcerbleeding in his twenties and had no other medical history related to bleeding or thrombosis. The patient's peripheral blood specimen was sent to the Seoul National University Hospital (SNUH), Seoul, Korea for further analysis. The results of laboratoryworkup performed in SNUH are presented in Table 1: Prothrombintime (PT) and aPTT before preincubation were 12.2 and 177.9 s, respectively. Prolonged aPTT was corrected after a mixing study. Factors VIII, IX, and XII were within normal ranges, whereas factor XI was slightly decreased (Table 1).
To exclude immunologic causes of aPTT prolongation, lupus anticoagulant, anti-cardiolipin antibody, anti-β2GPI antibody, and anti-nuclear antibody were tested, which all turned out to be negative.Under suspicion of PK deficiency, aPTT was measured after preincubation with an aPTT reagent for 20 minutes. However,aPTT shortening was not observed: the re-tested aPTT was 169.7 seconds, and after preincubation it was 132 seconds. PK activity was of borderline deficiency and PK antigen level was within the reference range. Sanger sequencing of the KLKB1 gene revealed a normal sequence. On the other hand, HK activitywas below the detection limit, and HK antigen was very low (Table 1).
Sanger sequencing of the KNG1 gene (exons 1–11 and the flanking regions; NM_000893) revealed compound heterozygous variants c.488delG and c.1165C>T. A novel variant, c.488delG in exon 4, which resulted in a frameshift mutation (p.Gly163-Alafs*20), was assessed as a pathogenic variant based on the 2015 American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines: PVS1 (a frameshiftvariant that leads to a truncated protein), PM2 (absent from controls in the Exome Aggregation Consortium and Genome AggregationDatabase), and PP4 (highly specific patient phenotype) [4]. A nonsense mutation caused by c.1165C>T generates a premature stop codon at position 389 (p.Arg389*). The same variant has been reported in a patient with severe HK deficiency [5]. Thus, our patient was confirmed as having HK deficiency with compound heterozygous KNG1 variants. We could not performa family study because the patient was lost to follow up.
In HK deficiency, PK activity and factor XI have been reported to be decreased or normal [36]. In our case, PK activity was low, which was measured by the clotting method, and factor XI was reduced. Both PK and factor XI circulate bound to HK, which explains the decreased levels of PK and factor XI in our patient.
Patients with HK deficiency are mostly asymptomatic, with prolonged aPTT. Despite their role in triggering the coagulation pathway, deficiency in any contact activation system components, including HK, does not lead to bleeding [1]. A few cases of HK deficiency with thrombosis have been reported: left vertebral basilar artery thromboses, deep vein thrombosis with pulmonary embolism, and splenic infarction [378]. However, the associationbetween HK deficiency and thrombosis has not been clarified.Deletion of murine kininogen gene 1 (mKng1) delayed thrombosisin an arterial injury model [9]. A recent study using a murinemodel revealed that HK deficiency protects mice from ischemicneurodegeneration [10]. In our case, the patient did not have any thrombosis-related medical history. Because of conflictingresults and limited data on the relationship between HK deficiency and thrombosis, we suggest that in cases of HK deficiencywith thrombosis, other causes of thrombosis should be thoroughly investigated to clarify whether the finding is coincidental,and thrombosis formation should be closely followed up.
 XML Download
XML Download