

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Syringocystadenocarcinoma papilliferum (SCACP) is a rare neoplasm arising from the apocrine glands or pluripotent appendageal glands1. In the World Health Organization classification of skin tumors, SCACP is listed as a malignant form of syringocystadenoma papilliferum (SCAP)2. Limited cases of SCACP have been reported, and it can be divided into invasive SCACP and SCACP in situ, which resembles other cutaneous malignant neoplasms. SCACP in situ is a rare variant form of adenocarcinoma in situ, which can accompany a pagetoid epidermal involvement3. SCACP can arise in any lesion of the body, although the head and neck are the most preferred areas. Most of the SCACPs are believed to arise from a pre-existing SCAP through a malignant transformation, while some were reported to be associated with nevus sebaceous through multistep progression4. Here, we report a rare case of SCACP and a review of other relevant literature.
Go to : 
CASE REPORT
An 85-year-old female visited our department owing to a painful mass on her perianal area. The patient recalled that the lesion was observed 1 year ago and had since increased in size.
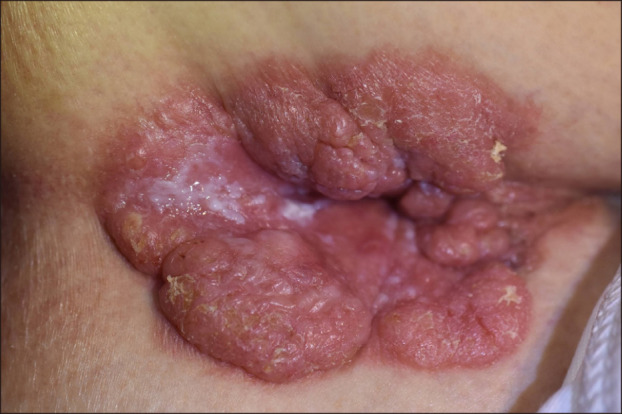
She had a history of hypertension, angina, and undiagnosed pulmonary nodule detected 3 years ago. Physical examination revealed a 7.0 cm×3.5 cm exophytic erythematous nodule, some of which was covered with whitish discharge and crusts (Fig. 1). Histological examination of the specimen from the perianal area showed epidermal invagination and papillary projections with a two-layered epithelium and stromal plasma cells. Tumor cells forming tubular structures showed nuclear atypia and mitotic cells. Pagetoid cells were observed in the epidermis (Fig. 2). The specimens showed positive results for periodic acid Schiff stain, cytokeratin (CK) 7, epithelial membrane antigen, carcinoembryonic antigen (CEA), and CK5/6, and negative results for p63 and gross cystic disease fluid protein-15. The proliferation marker (Ki-67) was present in more than 50 percent of the cells. Laboratory studies including complete blood count and routine chemistry showed normal values. Tumor markers such as total prostate-specific antigen, alpha-fetoprotein, CEA, and carbohydrate antigen 19-9 were also within the normal range. Imaging studies including abdominal computed tomography and fluorodeoxyglucosepositron emission tomography were performed to investigate locoregional or distant metastasis, and no other malignancy or metastasis besides the primary perianal skin tumor or pre-existing lung nodule was observed. Based on these findings, the perianal lesion was finally diagnosed as SCACP in situ. The patient initially underwent radiation therapy, as her family members denied surgery. Subsequently, the tumor size decreased, and no local progression was observed during the 8-month follow-up period after radiation therapy. We received the patient's consent form about publishing all photographic materials.
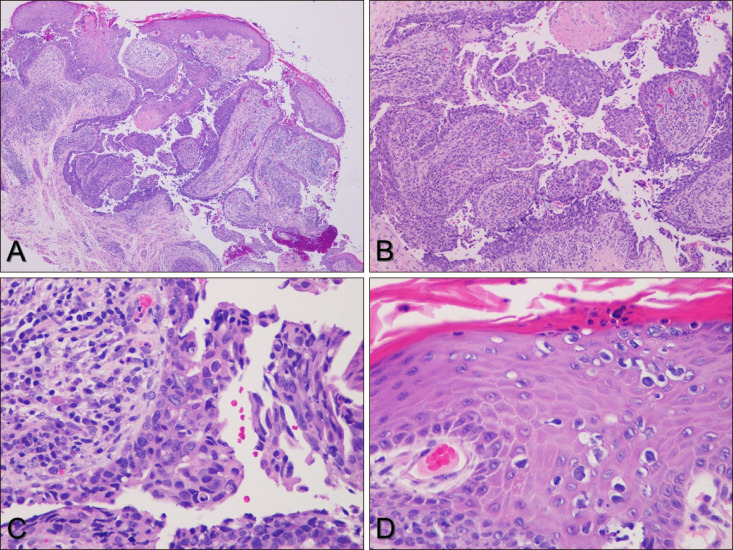 | Fig. 2Biopsy from perianal area. (A) Tumor shows tubular and cystic configuration (H&E, ×40). (B) Wall of cystic structure shows papillomatous projections into cavity (H&E, ×100). (C) Tumor cells forming tubular structures show pale cytoplasm and large atypical nucleus with atypical mitoses. Stroma of papillae contains plasma cells and lymphocytes (H&E, ×400). (D) Tumor cells in epidermis show pagetoid spreads (H&E, ×400).
|
Go to :
DISCUSSION
Thus far, to the best of our knowledge, only 49 cases of SCACP have been reported in the English literature. Based on our analysis, including our case, SCACP is common in old age, and the median age of the patients has been reported to be 63.6 (range, 22~93) years (Table 1)123456789101112131415161718. Although previous reports show no gender predilection1, the male:female ratio of the patients is 1.5 : 1. Clinically, it presents as a flesh-colored to hyperpigmented exophytic nodule, inflammatory plaque, or tumor. The overlying crust caused by the decapitation secretion of the apocrine gland is its characteristic feature5. The mean maximum diameter of the tumor is 3.98 cm (range, 0.4~16 cm). Out of the 50 reported cases (including the current case) of SCACP, the tumors were located in the head and neck area in 27 cases. Indeed, SCACP is often associated with SCAP, which is well known to have a preference for the head and neck area419. Perianal area is an uncommon site of SCACP, and only four cases involving SCACP in the perianal area, including our case, have been reported678. The occurrence of SCAP in nevus sebaceus of Jadassohn (NSJ) is well documented4. Some authors recently discovered that NSJ is a mosaic RASopathy, a mosaic mutation of HRAS and KRAS genes with activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)-Akt signaling pathways420. However, as observed in the current case, some SCACPs can arise in the trunk, axilla, genitalia, and extremities, and are not associated with a previous benign tumor. This suggests that SCACP can develop de novo. Owing to the limited reported cases of SCACP, its further pathophysiology should be revealed in future studies.
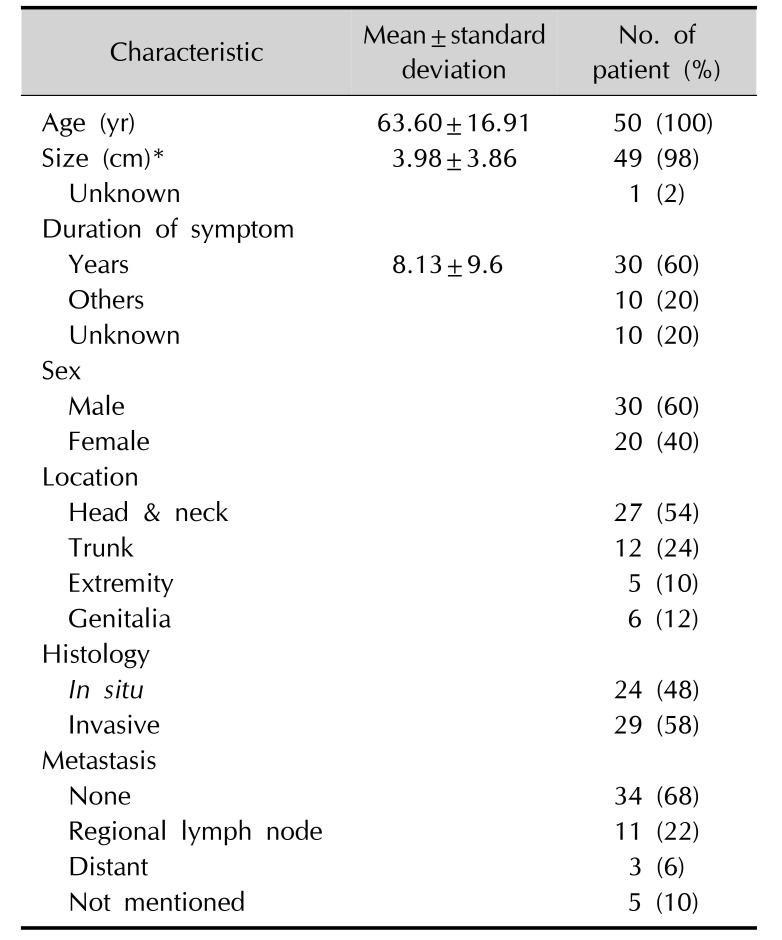
Table 1
![]()
Histopathologically, SCACP resembles SCAP in that it consists of a cystic and papillary growth of epithelial elements composed of an inner columnar and outer cuboidal layer, projecting downwards into the dermis. The inner columnar cell layer shows evidence of decapitation secretion.
The stroma of the tumor are infiltrated with inflammatory cells, especially plasma cells19. Unlike SCAP, SCACP presents different architectural and cytological features. The tumor in SCACP is asymmetric and poorly circumscribed, often extending deep into the subcutaneous fat. Multiple layers of invagination are often observed in SCACP. The common cytological features of malignant cells such as higher nuclear cytoplasmic ratio, nuclear irregularity, coarse chromatin, or increased mitotic activity are also observed in SCACP35910.
Histologically, SCACP can present as (1) SCACP in situ, (2) invasive SCACP, and, rarely, (3) invasive squamous cell carcinoma (SCC), characterized by the presence of invasive SCC component in the SCAP background4. Some other studies have reported that the histological features of SCACP include psammoma bodies, perineural invasion, pagetoid spread, squamous differentiation, and even sarcomatous change4611. There is no specific immunohistochemical marker that defines the diagnosis of SCACP. However, positive results of cytokeratin 5/6 (CK5/6), p63, and D2-40 staining tend to be associated with cutaneous neoplasms instead of metastatic adenocarcinoma, even though they are not entirely specific5810. In our case, the patient had a history of a lung nodule for the last 3 years, which was undiagnosed because of lack of biopsy. However, connection with the epidermis, presence of an in situ component, and positive results for in CK5/6 and CK7 in luminal cells can help distinguish primary adnexal tumor from metastatic adenocarcinoma.
Clinically, tumors on the perianal area should be differentiated from other diseases such as SCC, Bowen's disease, basal cell carcinoma, eccrine porocarcinoma, and malignant melanoma6. Histologically, hidradenoma papilliferum (HAP), extramammary Paget's disease (EMPD), cutaneous metastases from breast or gastrointestinal cancers, metastatic thyroid carcinoma, apocrine adenocarcinoma, and adenocarcinoma of the anogenital mammary-like glands should be differentiated6712. HAP is the most common adnexal skin tumor occurring in the perianal region, and it usually presents a more complex papillary growth pattern as well as absence of plasma cells within the stroma. EMPD can present with glandular structures. Cutaneous metastases from breast or gastrointestinal cancers lack papillary structures containing deep invaginations.
Although very few cases of SCACP have been reported, most of the patients have been surgically treated with wide local excision, which is considered as the standard treatment. In cases of inoperable disease or in patients who refuse surgery, chemotherapy or radiation therapy can also achieve good tumor control13. Among the 50 reported cases of SCACP, locoregional lymphatic metastases were found in 11 patients (22%) and distant metastasis were found in 3 patients (6%) (Table 1); all the 3 patients with distant metastasis died owing to disseminated disease. Although SCACP was thought to be a low-grade malignancy with a favorable outcome1314, the actual prognosis was not as good as that reported in previous studies. In conclusion, SCACP is an extremely rare adnexal tumor, and its definite pathophysiology has not been revealed. In most cases, SCACP is associated with SCAP. Multiple malignant progression from NSJ is one of the reliable hypotheses. Differential diagnosis of SCACP from other metastatic carcinomas is important, and patients should undergo careful history taking, physical examination, and immunohistochemical studies. Thus far, although SCACP was considered as a favorable disease, recent case reports showed that locoregional lymph node metastases and distant metastases are not rare. Complete surgical excision is the treatment of choice for SCACP, and chemotherapy or radiation therapy can also be considered in inoperable cases. Here, we report a rare case of SCACP with a review of other relevant literature. Further studies on a large number of cases are warranted.
Go to :
 XML Download
XML Download