

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Propionic acidemia (PA) is a severe metabolic disease that occurs in infants and children, which is inherited as an autosomal recessive pattern. The global incidence of PA has been estimated to range from 1:250,000 in Germany to 1:17,400 in Japan.1 In Korea, the incidence of organic acidosis is 1:8,000, but its prevalence is 0.07–0.19 per 100,000 population.2 Most cases are diagnosed during the neonatal period. The patient reported herein is a 4-year-old boy who did not have any symptoms associated with a metabolic disease. The patient was admitted owing to recurrent pancreatitis, and metabolic crisis occurred while receiving total parenteral nutrition (TPN). He was diagnosed and treated for PA and eventually showed full recovery.
CASE DESCRIPTION
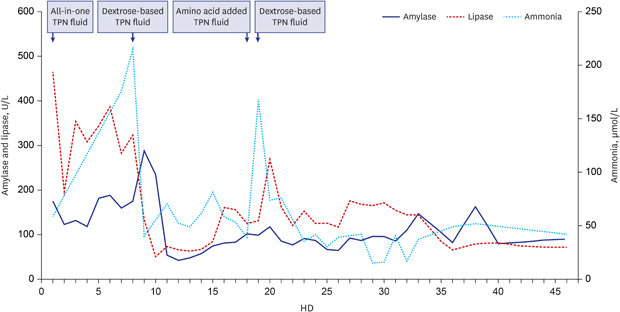
On January 27, 2014, a 4-year-old boy was admitted owing to a second episode of pancreatitis at a general hospital and experienced abdominal pain and vomiting for 3 days. The first episode of acute pancreatitis occurred 1 year before. He was referred to our children's hospital for further work-up and treatment. At arrival, he was fully conscious, and his symptoms were much improved. Laboratory tests revealed a hemoglobin level of 13.2 g/dL, white blood cell count of 8,720/µL, and platelet count of 334,000/µL. Amylase and lipase levels were 175 and 465 U/L (normal range, 22–80 and 0–60 U/L) respectively, which were improved from 773 and 1,969 U/L at a regional hospital 2 days prior. Coagulation tests revealed normal. He had metabolic acidosis with HCO3− level of 8 mmol/L, and a base deficit of 16 mEq. Urinalysis revealed severe ketonuria. Fasting blood glucose level was 65 mg/dL, and ammonia level was 59 μmol/L. Magnetic resonance cholangiography revealed a diffusely swollen pancreas without parenchymal necrosis and duct anomaly (Fig. 1). The patient received TPN consisting of all macronutrients, including amino acids. However, metabolic acidosis, intermittent vomiting, and decreased activity persisted. On the seventh day of hospitalization, he was transferred to the intensive care unit because of a sudden metabolic crisis with generalized tonic convulsion lasting for 2 minutes, with prolonged postictal drowsiness. His blood pressure decreased to 81/42 mmHg. Ammonia increased to 175 μmol/L. Amylase and lipase levels were 168 and 413 U/L, respectively. Platelet count was 86,000/μL, and PT and aPTT were not measurable (Fig. 2). Fibrinogen and antithrombin levels decreased to 114.5 mg/dL and 68.4%, respectively. D-dimer level increased to 1.6 mg/L, and disseminated intravascular coagulation was evident. Azotemia was not observed, and calcium decreased to 5.7 mg/dL.
Fig. 1
Magnetic resonance cholangiography revealed a diffusely swollen pancreas without parenchymal necrosis and duct anomaly.

Fig. 2
Level of amylase, lipase and ammonia during the admission. In HD1, TPN was started consisting of all macronutrients, including dextrose, amino acids, and fat emulsions. In HD8, all-in-one TPN fluid was changed to dextrose-based TPN fluid without amino acid. In HD18, we added amino acid in TPN fluid as a trial.
HD = hospital day, TPN = total parenteral nutrition.

The CT scan of brain was unremarkable. The chest X-ray showed signs of pulmonary edema, and the patient's oxygen saturation decreased to < 90%. The electrocardiogram showed tachycardia at a rate of 150 beats/min, and the QTc interval prolonged to 0.527 seconds. The echocardiogram revealed decreased left ventricular contractility, with an ejection fraction of 41.0% and fractional shortening of 19.8%, and digoxin was administered to treat toxic myocarditis. Intensive care was applied for myocarditis, hyperammonemia and disseminated intravascular coagulation caused by metabolic crisis. All-in-one TPN fluid was changed to TPN fluid without amino acid. In the metabolic disease work-up, tandem mass spectrometry showed an increase in C3 carnitine, indicating the possibility of PA and methylmalonic acidemia. Increased glycine level in plasma amino acid testing and increased 3-hydroxypropionic acid, methylcitric acid, and propyl glycine levels in urine organic acid testing suggest PA. Molecular genetic analysis revealed compound novel heterozygous mutations of PCCB gene: c.1316A>G (p.Tyr439Cys) and exon 8 deletion. Both mutations were predicted to be deleterious by in silico analysis database, SIFT, PolyPhen-2 and Mutation Taster. The patient's condition and all laboratory test results improved to reach the normal values after protein administration was stopped and replaced with supportive care, including methylmalonic propionic acidemia (MPA) formula, metronidazole, and antioxidant cocktail including biotin, vitamin E, L-carnitine. Five years of follow-up until now, the patient experienced several additional episodes of recurrent pancreatitis even after the administration of MPA formula and restriction of protein supplementation. After regular prophylactic use of metronidazole (weekly dose every month), no incidence of pancreatitis has occurred. Growth and development of the patient occurred normally.
DISCUSSION
PA is a metabolism disorder of propiogenic amino acids and odd-numbered fatty acid caused by a deficiency of mitochondrial propionyl-coenzyme A carboxylase (PCC). Most cases of PA are diagnosed within the first few days of life. Similar to a majority of congenital metabolic diseases, PA is associated with specific symptoms, such as vomiting, hypotonia, hyperventilation, and anorexia, accompanied by excessive acidosis during the neonatal period (71%–92%).3-5 On the other hand, late-onset PA, which manifests after 3 months of age, is characterized by less severe symptoms as well as a different pattern of symptoms. Late-onset PA usually involves vague symptoms, including anorexia, poor growth, poor nutrition, and neuromotor and psychological disabilities, which make the diagnosis difficult, often leading to a delayed diagnosis.5
Complications affecting the multi-organ system have been reported, and the severity of symptoms varies from mild to death.6,7 The most common complication associated with the gastrointestinal system is pancreatitis, and pancreatitis also occurs in association with various types of organic acidemia other than PA.8,9 The incidence of pancreatitis among PA patients has been reported to range between 4% and 18%.7,10,11 Acute pancreatitis among PA patients generally represents just one component of multiple organ failure that is detected at follow-up conducted during their entire life. Additionally, recurrent pancreatitis among PA patients is uncommon, and there have been no reports of chronic pancreatitis associated with PA.
In the meanwhile, this patient was not previously diagnosed with a metabolic disease, including during the treatment of recurrent bouts of pancreatitis. PA was found incidentally during TPN administration. The patient was in a metabolic decompensation state caused by acute pancreatitis. Furthermore, excessive propiogenic amino acids and odd-numbered fatty acids were administered intravenously, and the catastrophic complication of multiple-organ decompensation suddenly arose, which progressed to manifest as metabolic crisis and shock. Unfortunately, screening of organic acidemia is not included in government-subsidized newborn screening tests for inherited metabolic disorders.
The pathophysiology of PA in association with pancreatitis remains unclear. There are several possible mechanisms by which PA patients develop pancreatitis. In some studies, both the clinical and biochemical manifestations of pancreatitis persisted longer among PA patients than the other manifestations of pancreatitis.12,13 Metabolic derangements affect mitochondrial enzyme function.14 The accumulation of non-metabolized odd-chain fatty acids makes hypertriglyceridemia a risk factor for pancreatitis.15 Moreover, a lack of PCC—an acidic byproduct that causes cellular acidosis—activates pancreatic acinar cells to cause pancreatitis.16 Additionally, mitochondrial dysfunction causes acinar cell calcium elevation, which further facilitates pancreatitis development.17 Furthermore, inappropriate sites of pancreatic enzyme activation; deficiencies in methionine, carnitine, and antioxidants; and excess free radical production also lead to inflammation of the pancreas.18,19
If acute pancreatitis is diagnosed in PA patients, TPN can be safely administered if necessary, provided that the amount of protein administered does not exceed 1.5–3.5 g/kg body weight/day, depending on the age of a patient.20 However, the amount of protein should be individualized according to laboratory findings. Restriction of protein administration in children with PA may result in poor growth or developmental delay. MPA formula can be used for protein supplement, which has low or no methionine, threonine, valine, and isoleucine content.
In conclusion, although rare, children with PA presenting with vomiting or abdominal pain should be evaluated to rule out acute pancreatitis. Additionally, even among children who have never been diagnosed with a metabolic disease, if a child has previously experienced recurrent pancreatitis, metabolic pancreatitis caused by organic acidemia should be considered for differential diagnosis. Remarkably, both c.1316A>G (p.Tyr439Cys) and exon 8 deletion in PCCB gene have never been identified in PA patients before.
 XML Download
XML Download