

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Reversible reactions are commonly encountered in pharmacology, chemistry, biology, chemical engineering, etc.[123456] The influence of reversible metabolism on the pharmacokinetics of a drug and its metabolites has been the subject of considerable recent research.[567891011] An interesting example is the administration of chiral drugs. Two enantiomers of a chiral drug, such as ibuprofen[10] and propranolol,[11] often have different potencies and pharmacokinetic profiles.[891011] However, one enantiomer is usually interconverted into the other within the human body.[7891011] When a chiral drug is administered as a racemic mixture, its bioavailability and efficacy can be incorrectly estimated if reversible reactions between two enantiomers are not taken into account.[56] For the determination of an appropriate dosage regimen of a drug, it is therefore important to understand the characteristics of the reversible reaction between a drug and its metabolite.
In this tutorial, we focus on the basics of the pharmacokinetics of reversible metabolism. For a simple reversible reaction scheme, we use the matrix method to get exact solutions and plot them to find characteristic relationships between the pharmacokinetic profiles of a parent drug and its metabolites. In addition, we describe two approximation approaches to deal with complex problems and examine the conditions required for the successful application of each approach.
Go to : 
Theoretical analysis
Exact solution for the kinetics of reversible reaction
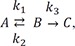
Where the first step is reversible with a forward rate constant k1 and reverse rate constant k2, and the second step is irreversible with the rate constant k3. The rate constant has the units of the amount per unit time, which are omitted for simplicity in this tutorial. The rate equations are:
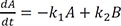
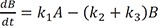
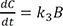
Linear ordinary differential equations can be solved using Laplace transformation or the matrix method.[23712] We used the latter to obtain an exact solution because of its simplicity. The above expressions can be written in matrix form:
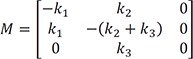
By setting
|−k1−λk20k1−(k2+k3)−λ00k3−λ|=0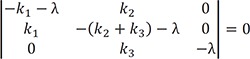 , where λ is the eigenvalue, and solving for λ, we get the following characteristic polynomial:
, where λ is the eigenvalue, and solving for λ, we get the following characteristic polynomial:
, where λ is the eigenvalue, and solving for λ, we get the following characteristic polynomial: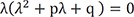
Where p = k1 + k2 + k3 and q = k1k3. The solutions of the quadratic equation in Eq. (5) are given by
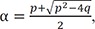
and
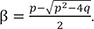
The resulting eigenvalues are 0, −α, and −β. These eigenvalues are substituted into the eigenvalue equation to yield the corresponding eigenvectors of
[001], [β−k1k1β], and [α−k1k1α]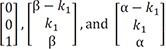 , spectively.
, spectively.
, spectively.Because the matrix M has three distinct real eigenvalues and their corresponding eigenvectors, we can express the solution as
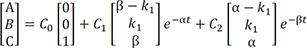
At t = 0, A = A0 and B = C = 0, and A + B + C = A0 at all times. Using these boundary conditions, we can solve Eq. (6) to get C0 = A0, C1 = −C2 and C1 = A0/(β – α). Then, the final expressions for the above differential equations (1–3) are
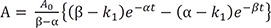
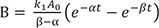
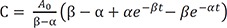
We can compare these results to those for a consecutive two-step reaction, as described in the previous tutorial.[13] We can easily show that the latter has the eigenvalues of 0, −k1, and −k3, by setting k2 = 0 in Eq. (5). The solutions for B and C, equations 8 and 9 in this study, are very similar to those for the consecutive reaction (Equations 7 and 9 in Reference,[13] respectively). Thus, the expressions for the maximum amount of B (Bmax) and the time to reach the peak (tmax) can be obtained in a similar way:
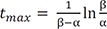
and
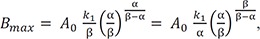
respectively.
Graphical insights into the kinetics of reversible reactions
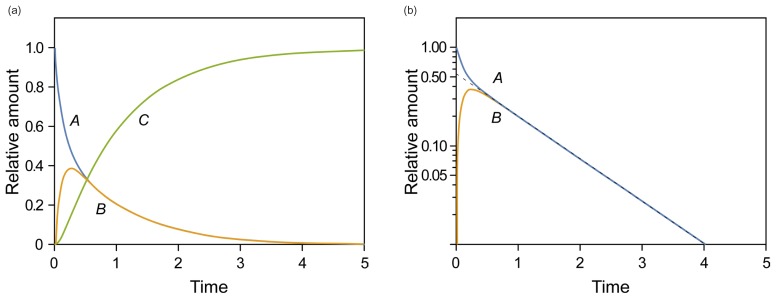
Figure 1 shows representative amount versus time curves of A, B, and C, plotted on a linear and semi-logarithmic scale for three different cases: (a) k1 = 10, k2 = 0.1, and k3 = 1; (b) k1 = 5, k2 = 4, and k3 = 2; (c) k1 = 1, k2 = 5, and k3 = 0.2. When k1 and k3 are relatively larger than k2, that is, the reverse reaction is negligible, linear plots of the amount versus time curves of A and B for the reversible reaction (Fig. 1a) look very similar to those for a consecutive irreversible reaction (see Figure 2 in Reference[13]). The semi-logarithmic plots for the former case (Fig. 1b), however, look different from those for the latter case (see Figure 3a in Reference[13]). In the former case, two curves are always parallel to each other in the elimination phase, whereas they are not in the latter case. The difference can be explained as follows. For an irreversible reaction, the amount of A is described by one exponential term, whereas for the reversible reaction, it is described by two exponential terms. For the latter, the first term in Eq. (7) or (8) decreases faster than the second term over time, because α is always larger than β. This means that the elimination phase is governed by the second term. Therefore, the first term can be neglected when t is sufficiently large. In semi-logarithmic plots of A and B, thus, both slopes for the elimination phase are linear and identical and given by –β/2.303. The y-intercepts for A and B are α−k1α−βA0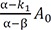 and k1α−βA0
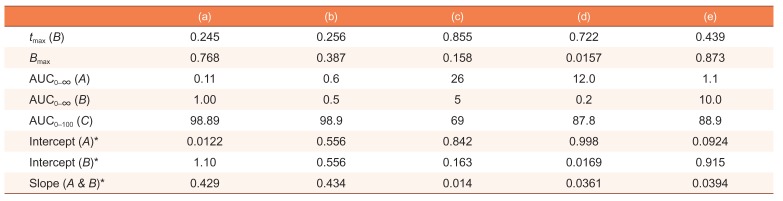
and k1α−βA0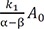 , respectively. When 2k1 = 2k2 + k3, the intercepts are the same, as shown in Figure 2. If 2k1 < 2k2 + k3, the curves A and B will not cross each other. As a third case, we set k1 = 1, k2 = 5, and k3 = 0.2, which satisfies this condition. It is noteworthy that the semi-logarithmic plots for this case (Fig. 3b) strongly resemble those for the consecutive irreversible reaction (see Figure 3b in Reference[13]). In the latter case, the semi-logarithmic plots of plasma concentrations versus time of A and B become parallel straight lines at large t only when the formation rate of B is much slower than its elimination rate; that is, the metabolism is formation rate-limited (FRL).[131415] In addition, the intercept for [B] becomes greater than that for [A] when the distribution volume of B, VB, is much smaller than that of A, VA (see Figure 3c in Reference[13]). However, we need to be cautious when we apply this feature to the interpretation of the time profiles of the semi-logarithmic plots of a drug and its metabolite because this feature is commonly observed in reversible metabolic reactions. The pharmacokinetic parameters for each case, including tmax and Bmax, are summarized in Table 1.
, respectively. When 2k1 = 2k2 + k3, the intercepts are the same, as shown in Figure 2. If 2k1 < 2k2 + k3, the curves A and B will not cross each other. As a third case, we set k1 = 1, k2 = 5, and k3 = 0.2, which satisfies this condition. It is noteworthy that the semi-logarithmic plots for this case (Fig. 3b) strongly resemble those for the consecutive irreversible reaction (see Figure 3b in Reference[13]). In the latter case, the semi-logarithmic plots of plasma concentrations versus time of A and B become parallel straight lines at large t only when the formation rate of B is much slower than its elimination rate; that is, the metabolism is formation rate-limited (FRL).[131415] In addition, the intercept for [B] becomes greater than that for [A] when the distribution volume of B, VB, is much smaller than that of A, VA (see Figure 3c in Reference[13]). However, we need to be cautious when we apply this feature to the interpretation of the time profiles of the semi-logarithmic plots of a drug and its metabolite because this feature is commonly observed in reversible metabolic reactions. The pharmacokinetic parameters for each case, including tmax and Bmax, are summarized in Table 1.
and k1α−βA0, respectively. When 2k1 = 2k2 + k3, the intercepts are the same, as shown in Figure 2. If 2k1 < 2k2 + k3, the curves A and B will not cross each other. As a third case, we set k1 = 1, k2 = 5, and k3 = 0.2, which satisfies this condition. It is noteworthy that the semi-logarithmic plots for this case (Fig. 3b) strongly resemble those for the consecutive irreversible reaction (see Figure 3b in Reference[13]). In the latter case, the semi-logarithmic plots of plasma concentrations versus time of A and B become parallel straight lines at large t only when the formation rate of B is much slower than its elimination rate; that is, the metabolism is formation rate-limited (FRL).[131415] In addition, the intercept for [B] becomes greater than that for [A] when the distribution volume of B, VB, is much smaller than that of A, VA (see Figure 3c in Reference[13]). However, we need to be cautious when we apply this feature to the interpretation of the time profiles of the semi-logarithmic plots of a drug and its metabolite because this feature is commonly observed in reversible metabolic reactions. The pharmacokinetic parameters for each case, including tmax and Bmax, are summarized in Table 1.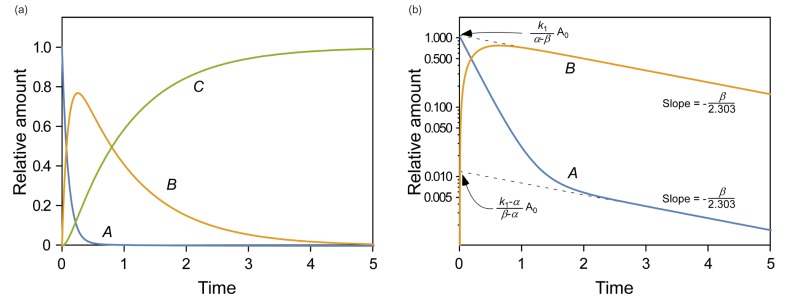 | Figure 1(a) Linear and (b) semi-logarithmic plots of the amount-time curves of A, B, or C in the reversible reaction, |
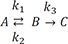
 | Figure 2(a) Linear and (b) semi-logarithmic plots of the amount-time curves of A, B, or C in the reversible reaction, |
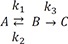
 | Figure 3(a) Linear and (b) semi-logarithmic plots of the amount-time curves of A, B, or C in the reversible reaction, |
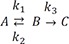
Table 1
Pharmacokinetic parameters for the following cases: (a) k1 = 10, k2 = 0.1, and k3 = 1 (as in Fig. 1); (b) k1 = 5, k2 = 4, and k3 = 2 (as in Figure 2); (c) k1 = 1, k2 = 5, and k3 = 0.2 (as in Figure 3); (d) k1 = 0.1, k2 = 1, and k3 = 5, (as in Figure 4a); (e) k1 = 10, k2 = 1, and k3 = 0.1 (as in Figure 4b)
![]()
The area under the curve (AUC) is an important pharmacokinetic parameter, representing total drug exposure over time. The AUC for A and B can be obtained by directly integrating Eqs. (7) and (8) from zero to infinity, respectively, and expressed as
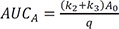
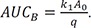
Thus, the ratio of AUCA to AUCB can be simplified to k2+k3k1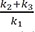 . The AUC for C can also be calculated by integrating Eq. (9) from zero to t and given by
. The AUC for C can also be calculated by integrating Eq. (9) from zero to t and given by
. The AUC for C can also be calculated by integrating Eq. (9) from zero to t and given by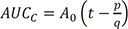
Using a time integration method,[1416] we can obtain the relationship between AUC and rate constant or clearance, which has the units of volume (V) per unit time (CL = k × V). By separating the variables in Eqs. (7–9) and integrating both sides with respect to their respective variables, we can write the equations as follows:
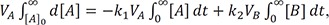
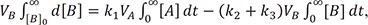
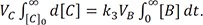
They should satisfy the following boundary conditions: [B] = [C] = 0 at t = 0; [A] = [B] = 0 and VC × [C] = A0 at t = ∞. By substituting k1 = CL1/VA, k2 = CL2/VB, and k3 = CL3/VC, we get
Eq. (19) can be rearranged as
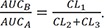
Approximation of the kinetics of reversible reactions
To get a good approximate solution for a reversible reaction scheme, approximation approaches have been developed and are widely used.[121920] Depending upon the reactivity of the intermediate species, we can use either of two approximation approaches. One is a steady-state approximation, in which the intermediate species B is very reactive and hence short lived. This approximation is useful for explaining reactions involving radical species. The other is an equilibrium approximation, in which B is quite stable and long-lived because the equilibrium reaction proceeds faster than the product formation reaction; that is, k1, k2 >> k3. For both approaches, the following condition should be satisfied:
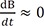
In the steady-state approximation, B is extremely unstable and very short-lived and thus is present in most of the time in extremely small amounts. By assuming that B ≈ 0, we can get a new boundary condition: A + C = A0. From Eq. (22), Eq. (2) can be rearranged to yield:
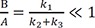
For the steady-state approximation, this ratio should be very small because the amount of B is much smaller than that of A. Substituting Eq. (23) into Eq. (1) and re-arranging it for A, we have
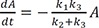
By separating the variables and integrating the resulting equation, we get
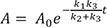
Using the above expression and the new boundary condition, A + C = A0, we obtain
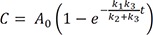
Now we can solve Eq. (2) for B using Eq. (26), to get
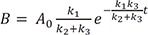
We plot the exact and approximate solutions for the case where k1 = 0.1, k2 = 1, and k3 = 5 (Fig. 4a). Because k1k2+k3=0.11+5~0.017≪1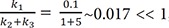 , the requirement for the steady-state approximation is satisfied. As the reaction begins, the amount of B quickly increases but is still extremely small (Bmax = 0.016 at tmax = 0.72) for most of the time, as expected. As shown in Figure 4a, the approximate solutions (dashed lines) agree well with the exact solutions (solid lines).
, the requirement for the steady-state approximation is satisfied. As the reaction begins, the amount of B quickly increases but is still extremely small (Bmax = 0.016 at tmax = 0.72) for most of the time, as expected. As shown in Figure 4a, the approximate solutions (dashed lines) agree well with the exact solutions (solid lines).
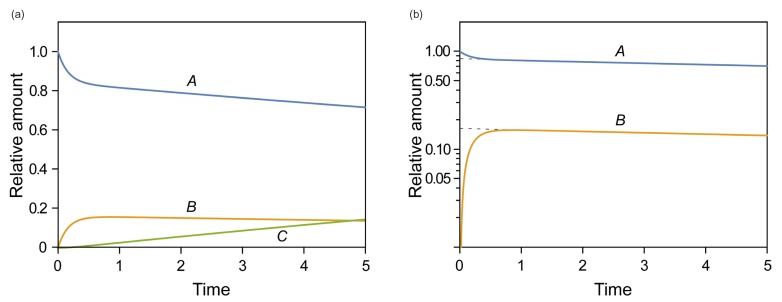
, the requirement for the steady-state approximation is satisfied. As the reaction begins, the amount of B quickly increases but is still extremely small (Bmax = 0.016 at tmax = 0.72) for most of the time, as expected. As shown in Figure 4a, the approximate solutions (dashed lines) agree well with the exact solutions (solid lines).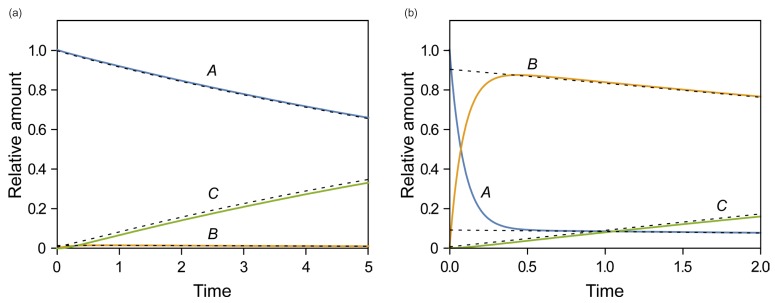 | Figure 4Linear plots of the amount-time curves of A, B, or C for the cases of (a) k1 = 0.1, k2 = 1, and k3 = 5, corresponding to the steady-state approximation, and (b) k1 = 10, k2 = 1, and k3 = 0.1, corresponding to the equilibrium approximation. The solid and dashed lines represent the exact and approximate solutions, respectively. The abscissa and ordinate denote time and the amount of each species, respectively, in arbitrary unit. See text for details.
|
For the equilibrium approximation, the reaction needs to be in pseudo-equilibrium. In such a case, we can express
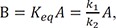
Where Keq is an equilibrium constant. Because
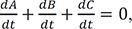
we also have
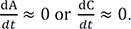
Because of the above condition, it is not feasible to directly solve Eqs. (1) and (2). Instead, we need to combine the two equations to get
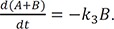
If we define X = A + B = A0 – C, from Eq. (28), we can express A and B as
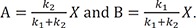
Now Eq. (30) can be solved for X to get
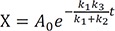
Then, we have
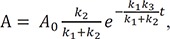
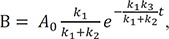
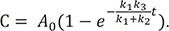
In Figure 4b, we show the plots of the exact and approximate solutions for the case where k1 = 10, k2 = 1, and k3 = 0.1. Unlike in Figure 4a, the amount of B is not negligible (Bmax = 0.87 at tmax = 0.44). The reaction rapidly reaches quasi-equilibrium, after which the amount of B slowly decreases. As shown in Figure 4b, the approximate solutions (dashed lines) agree well with the exact solutions (solid lines).
The difference between steady-state and equilibrium approximate approaches lies in the range of their applicability. [20] When we introduce an additional constraint, k2 >> k1 into the equilibrium approach and k2 >> k3 into the steady state approach, both methods will yield the same solution.[20] This relationship can be further explained by considering an approximate expression for β. Using the following binomial theorem
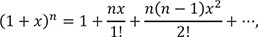
we can obtain the binomial expansion of β, which yields
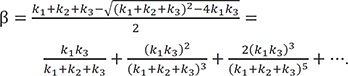
If k1 + k2 >> k3 or k2 + k3 >> k1, the second and higher order terms in the expansion can be negligible, and the first term can be further simplified to be the same as the exponent in the equilibrium approximation [see Eqs. (33–35)] or in the steady-state approximation [see Eqs. (25–27)], respectively. For a refined approximation, we can differentiate Eq. (23) for A and B with respect to time t to get
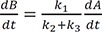
By substituting Eqs. (3), (23), and (37) into Eq. (29) and rearranging the resulting equation, we get
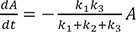
We expect a better approximate solution from the above equation compared with that produced by Eq. (24).
Application of the Reversible Reaction Scheme
The reversible reaction scheme is also used to describe drug distribution after rapid intravenous injection. Drug distribution within the body is often described by a two-compartment model with central and peripheral compartments.[21] In this model, an injected drug will be either eliminated or distributed within the central compartment, representing intravascular space, and the peripheral compartment, representing non-metabolizing body tissues. The distribution phase can be expressed by two linear differential equations similar to Eqs. 1 and 2.[21] The solution for drug concentration in each compartment can be obtained using a similar approach and described by two exponential terms.
To explain the kinetics of an enzyme-catalyzed reaction, a Michaelis-Menten model has been widely used.[222324] When an enzyme and a substrate form a complex, the binding of the substrate to the active site of the enzyme is non-covalent and reversible. If the conversion of the enzyme-substrate complex to a product is the rate-limiting process, the concentration of the complex can be considered to be constant. Then, the equilibrium approximation will give a hyperbolic relationship between the initial reaction rate, v_0, and substrate concentration [S], known as the Michaelis-Menten equation,
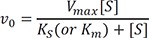
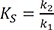
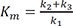
Go to :
Concluding remarks
We have described the mathematical approaches needed to determine the reaction kinetics of a reversible reaction system and provided graphical insights into the results. The knowledge acquired through this tutorial may help deepen the understanding of the pharmacokinetics of reversible metabolism and ultimately help with the prediction of the appropriate dose of a drug that is subject to equilibrium in a human body.
Go to :
 XML Download
XML Download