

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Glucose transporter type 1 (GLUT1), which is encoded by SLC2A1 (OMIM *138140) on chromosome 1, is a transmembrane transporter that allows glucose to pass through the blood-brain barrier. GLUT1 deficiency syndrome (OMIM #606777) is caused by mutations in SLC2A1, which result in hypoglycorrhachia and can limit glucose availability in the brain and lead to cerebral energy deficiency.1 Since the first description by De Vivo, et al.2 in 1991, there have been many reports on the wide spectrum of phenotypes of GLUT1 deficiency syndrome. This disorder is usually classified into two groups: 1) classical or typical GLUT1 deficiency, which involves infantile-onset pharmacoresistant epilepsy, intellectual disability, microcephaly, and complex movement disorders; and 2) nonclassical or atypical GLUT1 deficiency, which involves paroxysmal movement disorders, atypical childhood absence epilepsy, and myoclonic astatic epilepsy.134567 However, GLUT1 deficiency syndrome is not easily dichotomized, and is instead recognized as a continuum characterized by phenotypic diversity and evolution with age.148 Infantile-onset epilepsy can be alleviated during childhood, whereas movement disorders tend to emerge later, possibly due to changes in brain metabolism over time.8
Fortunately, GLUT1 deficiency syndrome has a specific treatment option—a ketogenic diet. Diet therapy is known to be effective for neurological dysfunctions, such as epilepsy and movement disorders. However, diagnosing GLUT1 deficiency syndrome can be challenging due to the variability in clinical features and age of onset, and phenotypic evolution over time. Although some authors have suggested diagnostic clues such as fluctuation associated with fasting or improvement with meals, in particular, self-induced high-fat diet,91011 it is not trivial to find these clues from the patients' recalls. The disease seems to be clinically underdiagnosed, even in the era of genomics.12 The aim of our study was to examine and present the pleiotropic and evolving phenotypes of nine patients with GLUT1 deficiency syndrome caused by mutations in SLC2A1.
Nine patients with pathogenic variants in SLC2A1, who visited the pediatric neurological clinic at Seoul National University Children's Hospital, were enrolled. We retrospectively reviewed their medical records. We collected detailed information about their prenatal and perinatal history, family history, age of symptom onset, initial symptoms, psychomotor development, neurological examination results, brain magnetic resonance imaging (MRI) findings, electroencephalography (EEG) findings, results of cerebrospinal fluid (CSF) studies, genetic tests, and treatment outcomes. This study was approved by the Seoul National University Hospital Institutional Review Board (IRB No. 1812-119-996), and blood samples were obtained from enrolled patients and their parents who provided informed consent. Identified variants through genetic testing were classified as pathogenic, based on the 2015 guidelines of American College of Medical Genetics and Genomics.13
The patients included eight boys and one girl (Table 1). The mean age at disease onset was 9.1 months (range, 1–24 months). The mean age at diagnosis was 9.4 years (range, 13 months to 26 years). Four patients (44%) initially manifested with seizures, four patients (44%) presented with developmental delay, and one patient presented with neonatal apnea. During the clinical course, all patients developed seizures of the mixed type, including absence seizures, generalized tonic–clonic seizures, generalized tonic seizures, myoclonic seizures, and focal seizures. Absence seizures and generalized tonic–clonic seizures were the most common. Seven patients had exhibited seizures before reaching 2 years of age, and two patients had developed epilepsy at the age of 7 and 11 years. Seizures were poorly controlled in most patients, and the average number of antiepileptic drugs was 3 (range, 1–5). Seven patients had medically intractable seizures with three or more antiepileptic drugs. Five patients (56%) exhibited movement disorders such as ataxia, dystonia, or dyskinesia over time (Fig. 1). Ataxia was relatively permanent, while dystonia or dyskinesia tended to be paroxysmal. The mean age of onset of movement symptoms was 5.1 years (range, 24 months to 10 years). The patients had varying severity of delayed psychomotor development or intellectual disability. Notably, only one patient had microcephaly. Other clinical features included oculogyric movements or apnea. Aggravating factors such as fasting, exercise, or fatigue were reported in four patients (44%), but no relieving or aggravating factors were identified in the other patients. Fluctuation of clinical symptoms along with the meals was recalled in only three patients (Patients 2, 5, and 7).
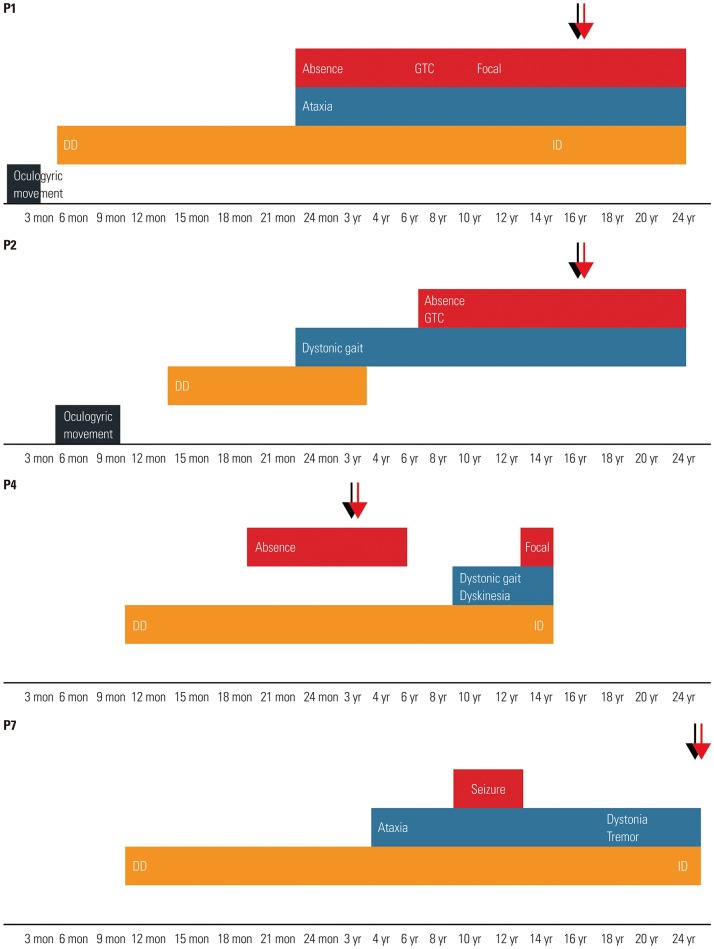

Fig. 1
The graph represents evolving phenotypes of four patients (Patients 1, 2, 4, and 7). Lower axis of the graph represents patient's age. Red and light blue represent epilepsy and movement disorders, respectively. Orange represents developmental delay (DD) and intellectual disability (ID). Black represents other atypical symptoms. Black arrows represent age at diagnosis, and red arrows denote age during treatment (ketogenic diet). GTC, generalized tonic–clonic.
![]()
Table 1
Clinical, Laboratory, Radiological, and Molecular Features of Patients with GLUT1 Deficiency Syndrome
| Patient No. | Sex | Age at onset: Dx:Current age or last f/u | Initial symptoms | Movement symptoms (onset age) | Global DD or ID (IQ, if available)/Epilepsy (onset age, sz type) | Microcephaly | Brain MRI | CSF-to-serum glucose ratio | Genotype | Inheritance, Reported [Ref.] | Pathogenicity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 6 mon:16 yr:19 yr | DD Ataxia | Ataxia (30 mon) | + (mild ID, IQ 50) /+ (2 yr, A-GTC-F) | - | N | 0.36 (34/94) | p.Val131Cysfs | De novo, novel | P |
| 2 | M | 18 mon:16 yr:18 yr | DD OM | Dystonia (24 mon) | +/+ (7 yr, A) | - | N | 0.36 (37/102) | c.276-1G>A | De novo, previous report [21] | P |
| 3 | M | 12 mon:13 yr:15 yr | Seizure | Dystonia (7 yr) | + (mild ID, IQ 54) /+ (12 mon) | - | Cerebellar atrophy | 0.4 (46/114) | p.Arg333Trp | From mother, previous report [22] | P |
| 4 | M | 12 mon:3 yr:16 yr | DD | Dyskinesia & dystonia (10 yr) | +/+ (20 mon, A-F) | - | N | 0.4 (39/97) | p.Phe434Ile | De novo, novel | P |
| 5 | M | 4 mon:7 yr: f/u lost after 7 yr 8 mon | Seizure | - | +/+ (4 mon, A-GTC) | - | Delayed M. | N/A | p.Arg153His | De novo, previous report [4] | P |
| 6 | M | 4 mon:17 mon: f/u lost after 3 yr 2 mon | Seizure | - | +/+ (4 mon, MC) | - | Delayed M. | N/A | p.Gly75Arg | De novo, novel | P |
| 7 | M | 24 mon:26 yr:28 yr | DD | Ataxia (4 yr) Dystonia (N/A) | +/+ (11 yr, N/A) | - | N | 0.36 (32/87) | p.Gln242* | Unknown, previous report [23] | P |
| 8 | M | 1 mon:18 mon: f/u lost after 18 mon | Apnea | - | +/+ (7 mon, MC-GT) | + | N | N/A | p.Arg126His | De novo, previous report [24] | P |
| 9 | F | 1 mon:13 mon: f/u lost after 21 mon | Seizure | - | +/+ (1 mon, F-GTC-GT) | - | N | N/A | p.Gly419Arg | De novo, novel | P |
No., number; Dx, diagnosis; f/u, follow-up; DD, developmental delay; ID, intellectual disability; IQ, intelligence quotient; sz, seizure; CSF, cerebrospinal fluid; M, male; F, female; mon, month(s); yr, year(s); OM, oculogyric movement; N/A, not available; A, absence; GTC, generalized tonic–clonic; F, focal; MC, myoclonic; GT, generalized tonic; N, normal; M., myelination; P, pathogenic.
![]()
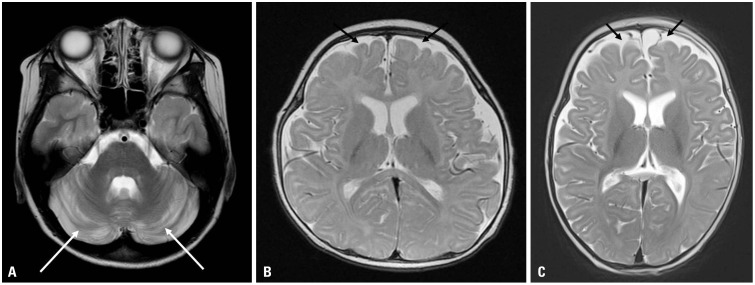
Brain MRI showed no abnormalities in six patients, cerebellar atrophy in one patient, and delayed myelination in two patients (Fig. 2). Interictal EEG was performed in nine patients with seizures, and showed generalized or focal epileptiform discharges with or without background slowing. CSF glucose levels were checked in five patients whose parents gave consent for lumbar puncture. CSF glucose levels ranged from 32 to 46 mg/dL, and CSF-to-serum glucose ratio was <0.4. Ketogenic diet was applied in five patients (Patients 1–4 and 7), and was effective in controlling seizures and movement symptoms. All patients except one (Patient 4) maintained this diet therapy for >1 year.
Nine pathogenic variants were identified. All nine different mutations were identified in the nine patients, and four were novel mutations. Among the eight patients whose parental tests were performed, seven had de novo mutations, while one (Patient 3) had a pathogenic variant inherited from his similarly affected mother. The variants were scattered across all exons (Supplementary Fig. 1, only online) without hot spots. Although about half of the variants had been identified through targeted single-gene analysis under the clinical suspicion of GLUT1 deficiency syndrome, the other half were found via epileptic encephalopathy panel sequencing (Patients 6 and 9) or whole-exome sequencing (Patients 1, 2, and 8).
GLUT1 deficiency syndrome is a treatable neurometabolic disorder, for which early diagnosis and treatment are very important. However, due to its nonspecific, overlapping, and evolving symptoms during development, this disorder remains underdiagnosed.412 The diversity of phenotypic features, severity, and clinical course, along with the phenotypic evolution with age mean that an early diagnosis can be challenging. A long diagnostic odyssey and variable, not uniform, clinical course have been reported in previous studies.3412 As in a recent study that reported a median age of diagnosis as 8 years 5 months,7 our study also revealed a substantial delay in the diagnosis of GLUT1 deficiency syndrome. In our study, the mean age of disease onset was 9.1 months, but the mean age of diagnosis was 9.4 years. Alter and colleagues reported that the earliest symptoms are dominated by seizures and other paroxysmal events, which are replaced by movement symptoms during adolescence.814 However, in our study, three out of five patients who were followed up to adolescence showed an atypical clinical evolution that differed from the typical course reported in previous studies. For example, Patients 2 and 7 showed movement abnormalities followed by epilepsy, and Patient 1 developed epilepsy and ataxia simultaneously. A high clinical suspicion and awareness, as well as recognition of phenotypic diversity and evolution over time, are very important for preventing a delayed diagnosis in patients who have seizures, neurodevelopmental problems, or associated abnormal movements with an unexplained etiology at any age.
The mechanism by which phenotypes vary or evolve with age is not clear. One possible explanation is that impaired glucose transmission to the brain does not affect prenatal or perinatal development. The brain glucose utilization rate increases from birth until about the age of 4 years, a time in which a child's cerebral cortex uses more than twice as much glucose as an adult's cortex; and this rate remains high until age 10, and then declines to reach the adult level at age 16–18 years.15 Therefore, the ontogeny of cerebral glucose metabolism is similar to the evolving pattern of epilepsy in GLUT1 deficiency syndrome.14
GLUT1 deficiency is representative of treatable neurometabolic disorders with intellectual disability. Since the first report by De Vivo, et al.2 in 1991, the effects of ketogenic diet have been supported, and the recently modified Atkins diet has also been known to be effective.1617 Controversy remains on whether diet therapy is highly effective for treating movement symptoms and cognitive impairment, as well as seizures.121416171819 Four of our patients (Patients 1–3 and 7) started diet therapy in adolescence or adulthood, but they showed favorable outcomes for most of the symptoms, including seizures and movement symptoms, in addition to subjective improvement in cognitive function. Further studies are needed to assess the overall outcome of a ketogenic diet by initiation ages, treatment duration, and type of diet in patients with GLUT1 deficiency syndrome.202122 In addition, medications such as acetazolamide can be applied for paroxysmal movement symptoms for cases with diet therapy failures.23
Atypical initial manifestations can be an obstacle to the early diagnosis of GLUT1 deficiency syndrome. In our study, oculogyric movements during neonatal or early infancy were found in four patients (44%). Oculogyric movements or crisis are easily mistaken as strabismus, but can be a key feature of GLUT1 deficiency syndrome.12 Although microcephaly also emerges during infancy or early childhood in most patients with this syndrome, only one patient in our series had microcephaly.24 Some symptoms, such as absence seizures, went unnoticed but were later recognized through comprehensive history taking. Our study recapitulates the atypical clinical features and phenotypic evolution of GLUT1 deficiency syndrome. Although a clue-based assumption, followed by CSF analysis and/or targeted single-gene analysis, is the most efficient diagnostic pathway,91011 diagnosis in actual clinical practice remains challenging. As shown in this study, application of next-generation sequencing can be helpful in shortening the time needed for the diagnosis of disorder with phenotypic variability and evolution.
The present study also expands the genotypic spectrum of GLUT1 deficiency syndrome. Whereas missense mutations are often associated with milder symptoms, no clear-cut phenotype–genotype correlations have been established.4 Although about three-quarters of the patients in our study had missense mutations, they did not show a milder phenotype compared to other patients with truncating mutations. Our patients exhibited interindividual phenotypic variability even though the same mutations have been reported previously,425262728 which suggests the presence of genetic modifiers such as secondary genes. Therefore, the genotype does not always predict the phenotype.
In conclusion, GLUT1 deficiency syndrome should be considered in patients with typical phenotypic evolution, such as infantile-onset epilepsy followed by movement symptoms around childhood or adolescence, as well as in atypical patients with early onset movement disorders and/or late onset epilepsy, or even in those with nonspecific and unexplained intellectual disability. Biochemical analysis and recent advances in genomic technology have made it possible to establish a diagnosis efficiently. Early diagnosis that leads to prevention and treatment is important for the neurological improvement,816 as well as ending the long diagnostic odyssey and providing genetic counseling, in affected children and adults.
 XML Download
XML Download