

 PDF
PDF ePub
ePub Citation
Citation Print
Print



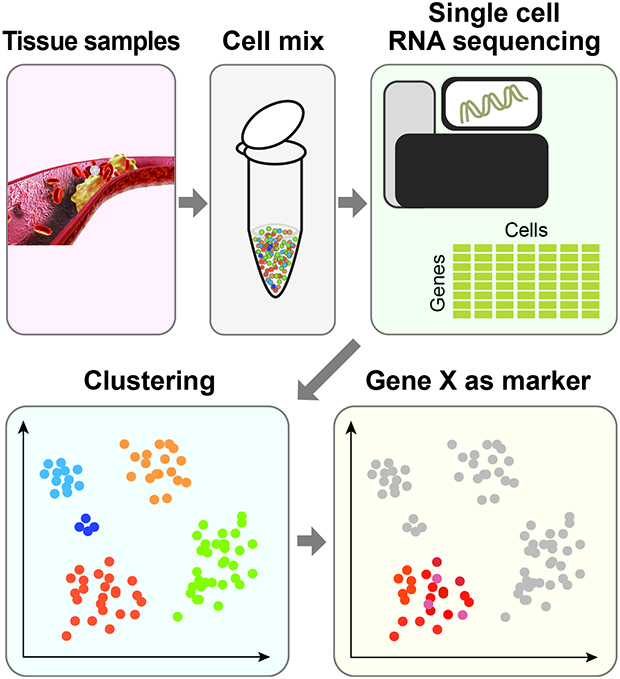
INTRODUCTION
Atherosclerosis is a chronic inflammatory disease driven by the interplay of many types of cells, including immune and stromal cells, with diverse phenotypic and transcriptomic changes.1234 A major limitation to the current transcriptomic methods, such as bulk RNA-sequencing (seq) analysis, is that they only capture transcriptional changes of the whole population rather than individual cells. Gene expression is heterogeneous, even in similar cell types.5 The stochastic nature of gene expression has a functional role and can lead to cell fate decisions.67 By measuring transcriptomic profiles at the single cell level, single cell RNA seq (scRNAseq) is an effective approach to deal with heterogeneous cell populations. The scRNAseq can detect the transcriptome of a rare cell population8 and study the trend of gene expression across the population of cells.9 The scRNAseq has been applied to various species, tissues (human and mouse), and studies to reveal cell-to-cell gene expression variability.10 Compared with the analysis of bulk RNAseq, which is mainly focused on identifying differentially expressed genes, scRNAseq provides various angles to study heterogeneity, cell interactions and transcriptomic changes along development or upon treatment. Algorithmic development has followed the new type of data generation. Therefore, the appropriate analytical approaches must be applied to effectively handle scRNAseq data.
In recent years, we have observed a number of atherosclerosis studies using scRNAseq.111213141516 In combination with various computational algorithms, the use of scRNAseq provided new information and knowledge about atherosclerosis, and it is expected that this new technology will become more popular. In this review, we aim to provide a guide about the scRNAseq technology used in recent studies of atherosclerosis. We will discuss the results from recent studies using scRNAseq in the field of atherosclerosis and introduce how the scRNAseq data were analyzed.
THE scRNAseq FOR THE STUDY OF ATHEROSCLEROSIS
The scRNAseq was first utilized in the field of atherosclerosis to investigate the driven plasticity of Forhead box P3+ T regulatory cells (Tregs) in an apolipoprotein E (ApoE) deficient mice model, a widely used system to study cardiovascular and respiratory diseases.11 Briefly, scRNAseq data were generated from 270 cells which were clustered into 3 groups: Treg, type 1 T helper (Th1)/Treg, and Th1. It has been known that atherosclerosis promotes the formation of an intermediately plastic Th1/Treg subset. Th scRNAseq data was used to confirm the results of flow cytometry about the existence of the previously uncharacterized Th1/Tregs group. The scRNAseq was further used to expand the list of genes that are differentially regulated in the Th1/Treg cell population. For instance, compared with Treg and Th1 cell populations, the Th1/Treg cell population is characterized by the downregulation of multiple Treg immunosuppressive genes, such as Tnfrsf4, Tnfrsf9, Tnfrsf18, Icos, Ctla4, and Treg-lineage transcription factors, such as Ikzf2, Ikzf4, and Foxp3.11
In 2018, Cochain et al.12 obtained transcriptome profiles of 372 control cells and 854 cells from diseased aortas. Among 13 aortic cell clusters, they identified 3 types of macrophages (resident-like, inflammatory and the previously uncharacterized triggering receptor expressed on myeloid cells 2 [TREM2+]). The scRNAseq was essential to identify TREM2+ macrophages, which do not belong to any of the previously known 2 types (M1- and M2-polarized) of macrophages. They found that the inflammatory macrophages and TREM2+ macrophages were almost exclusively observed in the cells from diseased aortas. In his study, Cochain et al.12 used the Seurat package (https://satijalab.org/seurat/)17 for clustering cells and identifying the marker genes associated with each cluster. Furthermore, they compared the number of diseased and control cells in each cluster.
In a similar study, Winkel et al.13 clustered 555 cells from control and 909 from disease cells and identified 11 leukocyte populations, including B cell subsets, using the Seurat package. Besides the cell subpopulation, they studied cell composition in the media, adventitia, and lesion and adventitia + tertiary lymphoid organs (ATLO). They calculated the composition of cells using the decomposition analysis based on bulk RNAseq and scRNAseq and reported that samples from lesions had a significantly smaller number of B cells compared to other samples.
More recently, Gu et al.14 also examined cell populations in normal and ApoE-deficient murine aortic adventitia. Besides clustering analysis to identify sub-populations, they studied cell communication between mesenchymal cells and macrophages in ApoE-deficient adventitia by evaluating the expression of ligand-receptor pairs.14 This analysis identified that chemokine (C-C motif) ligand 2, a chemokine secreted mainly by inflammatory cells and dysfunctional endothelial cells in atherosclerosis, was selectively expressed in a mesenchymal population for communication with macrophages.
Moreover, Kim et al.15 identified 11 leukocyte subpopulations, including diverse macrophage sub-clusters, in an atherosclerotic aorta using scRNAseq. These results were compared with the results from the foamy and non-foamy macrophage populations identified using flow cytometry.15 Another study investigated the features of monocyte-to-macrophage transition using scRNAseq in combination with genetic fate mapping of myeloid cells derived from CX3C chemokine receptor+ precursors during atherosclerosis progression and regression. They performed pseudo-time analysis after aligning cells along the pseudo-transition time.16
To generate scRNAseq data, Butcher et al.11 used Fluidigm C1. The C1 system enables size-based cell selection and is currently able to collect up to 800 cells. Other studies used the Chromium System (10× Genomics) in which a droplet-based microfluidic platform is used to sequence thousands of cells in parallel. A recent review paper summarized the commercially available instruments for single-cell collection.18
Because of its ability to measure the transcriptome at the single cell level, scRNAseq is increasingly used in various areas in biology. We aim to review some of the analyses made possible by scRNAseq output, which can be used for the study of atherosclerosis as well.
THE scRNAseq DATA ANALYSIS
1. Pre-processing
Raw data generated by a sequencing machine are processed to have read counts or number of molecules. The count information can be represented in a matrix which shows the gene expression across cells (Fig. 1A). To exclude low-quality information from scRNAseq, a series of quality controls (QC) are generally required. QC is employed to ensure that the quality of scRNAseq data is sufficient for subsequent downstream analysis. Common practices include removing cells with a low count depth or few detected genes. Some transcriptomic information can be from multiple cells (doublets). These unwanted doublets can be removed using doublet detection tools including Scrublet19 and DoubletFinder.20
 | Fig. 1The scRNAseq data processing for atherosclerosis. (A) scRNA procedure. The scRNAseq data are collected from each sample which are represented in a table. PCA and/or tSNE is applied to reduce the dimension for the sake of clustering. Genes expressed in each cluster are examined. (B) Clustering results using scRNAseq data from the study by Lin et al.16scRNAseq, single cell RNA sequencing; PCA, principal components analysis; tSNE, t-distributed stochastic neighbor embedding; Ear2, V-erbA-related protein 2; Irf7, interferon regulatory factor 7.
|
Once the matrix is obtained, normalization can be considered to compensate for cells with different numbers of barcodes or read depth. Normalization scales count data to a relative expression abundance. In addition, the batch effect should be removed when considering scRNAseq data from several sources. The batch effect is a common source of technical variation that can arise from various sources, including disparate cell dissociation protocol, library preparation, and sequencing platforms. Comprehensive benchmarking tests were applied for normalization and batch effect correction for the dataset for droplet-based scRNAseq data21 showing that pooling-based size factor estimation by Scran22 is one of the best working normalization approaches. The Seurat package is equipped with batch effect correction,17 but trying diverse approaches after considering the underlying biological processes is also recommended for batch effect correction.
2. Clustering
Clustering is a useful approach to identify various types of cells from scRNAseq. Hierarchical clustering has been widely used for bulk-cell RNAseq analysis. Butcher et al.11 used hierarchical clustering for 270 cells and identified 3 groups of cells. As the number of cells increases, the need for new approaches to handle high-dimensional scRNAseq data arises. Various algorithms for processing multi-dimensional scRNAseq data have been developed. Generally, these clustering methods rely on dimension reduction algorithms to avoid unnecessary noise (features). Dimensionality reduction approaches, such as principal components analysis (PCA), have been widely used to analyze both bulk RNA-seq data as well as scRNAseq data.1723 For instance, PCA allows the conversion of a higher dimensional dataset into a lower, often 2 or 3, dimensional dataset with more important and uncorrelated variables (dimensions) called principal components. Subsequently, cells can be clustered and envisioned in 2- or 3-dimensional (2D or 3D) space. Similarly, t-distributed stochastic neighbor embedding (tSNE) has also been successfully used to visualize cells in a reduced space.17 Moreover, tSNE is used to visualize cells in 2D or 3D space while reflecting true distances in the original space as far as possible so that cells of a particular cell type tend to be located nearby in 2D space. Finally, clustering is performed on cells by grouping them in the reduced space. Fig. 1B shows the tSNE-based analysis using the dataset by Lin et al.16 to understand macrophage heterogeneity during atherosclerosis progression and repression. From the tSNE plot, we can identify sub-clusters with V-erbA-related protein 2 (Ear2) expression (corresponds to RentnlahiEar2hi macrophage) and interferon (IFN) regulatory factor 7 (type 1 IFN signature) (Fig. 1B). In a similar manner, cell types are assigned based on a priori known marker genes. Cochain et al.12 identified cells, such as B cells (using Cd79a, Cd79b, Ly6d, and Mzb1), C-X-C chemokine receptor type (CXCR)6+ T cells (using CXCR6, Icos, Cd3g, and Il7R) and natural killer cells (using Klrb1c, Ncr1, Klra8, and Klrc1), based on the associated marker genes.
A number of algorithms have also been exclusively proposed for scRNAseq data analysis, including SC324 and SIMLR.25 CellBIC was designed to identify small cell subpopulations without losing information by dimension reduction.26 GiniClust has also been proposed to identify rare cell population8. Recent advances allow for ultra-fast clustering of more than 1 million cells.27
3. Cell composition comparison
One of the downstream applications of scRNAseq analysis is the comparison of cell compositions. For instance, Cochain et al.12 compared the number of cells from the control and the diseased aortas for each of the 3 clusters of macrophages and found that TREM2+ macrophages were almost exclusively observed in the cells from diseased aortas. In addition, the same quantitation could also provide an estimation of cell composition of bulk-cell RNAseq. This approach may be particularly useful when samples are collected from a different section of tissue. If scRNAseq is provided for a section (so that cell subpopulations are obtained), the cell composition of another section can be estimated from the bulk RNAseq using computational deconvolution based on scRNAseq28 (Fig. 2). Winkel et al.13 used CIBERSORT29 to perform deconvolution of cells using bulk-RNA-seq from the media, adventitia, lesion and adventitia + ATLO.
4. Pseudo-time analysis
When cells are represented in a lower dimensional space, those with similar transcriptomes will be located nearby on a plot, e.g. using tSNE. When cells are collected in different time stamps during differentiation, mature cells will be located far from progenitors, and cells being differentiated will be located in the middle. The path that links the cells can be regarded as a “pseudo” time9 (Fig. 3). This allows for longitudinal analysis of gene expression (e.g. development). Pseudo-time can be used to model transcriptomic changes during the development of atherosclerosis. Gene expressions can be analyzed along pseudo-time. For instance, the expression level of elastin deceases during direct cardiomyocyte conversion, while the expression level of troponin I1, slow skeletal type increases (Fig. 3). Furthermore, Lin et al.16 used the pseudo-time analysis along the fate-mapping during atherosclerosis progression and regression. This analysis found 53 genes significantly correlated with pseudo-time score, including CXCR4 and Ctsd. Monocle has been used for pseudo-time analysis.9 TSCAN combines clustering with pseudo-time analysis.30 Partition-based graph abstraction could be useful when complex trajectories are expected.31
 | Fig. 3Pseudo-time analysis using scRNAseq. The scRNAseq are obtained from cells during direct conversion to cardiomyocytes49 and reprocessed. Fibroblast cells are located on the left side and cardiomyocyte cells on the top right. Cells can be aligned in between based on their transcriptomic similarities. When aligned, pseudo-time analysis is applied. The expression level of Eln, a fibroblast marker, decreases along the pseudo-time.scRNAseq, single cell RNA sequencing; Eln, elastin; Dlk1, delta like non-canonical notch ligand 1; Tnni1, troponin I1, slow skeletal type; Tnni3, troponin I3, cardiac type.
|
5. Reconstruction of gene regulatory networks
Reverse engineering reconstructs gene regulatory networks from gene expression information.32 It usually requires a large amount of expression data. By providing transcriptomic information for each single cell, scRNAseq can be a good resource for reconstructing the regulatory networks. Pseudo-time has also been used to identify potential downstream target genes9 (Fig. 3). Software tools such as SCODE were developed to reconstruct gene regulatory networks from scRNAseq data.
6. Adding spatial information to scRNAseq
Another major limitation of current transcriptomic analysis workflow is that once the cells are isolated from tissue for scRNAseq, the cell location and orientation information is lost. To restore approximate location information, tissues can be mechanically sampled from different spots. For instance, Winkel et al.,13 used the spatial information by comparing cells from whole-atherosclerotic aortas versus aortic leukocytes. Another strategy has also been introduced in which barcoding the native tissue location has been proposed.33 This approach dissects the histological section with a grid, and each spot is barcoded to provide position information. Currently, a grid contains 10–30 cells depending on the tissue. The RNAs along with the position information are sequenced together. Spatial transcriptomics is now part of 10× Genomics (https://spatialtranscriptomics.com/). Another approach called MERFISH uses fluorescence in situ hybridization to provide spatial information in order to map the identity and location of specific cell types.34 Statistical test can be applied to identify genes whose spatial patterns are significantly different.35
7. Studying cell communications
The expression of ligand-receptor pairs can be utilized to study cell communication.36 Specifically, Gu et al.14 investigated the ligand-receptor interaction between cell subtypes by considering the transcriptomic levels of ligands and their corresponding receptors. In cells from the adventitia, this computational prediction showed the importance of mesenchyme populations for maintaining adventitial homeostasis. For instance, cells expressing Cd34 and Cav1 interacted with Sell and Icam1 expressed by inflammatory macrophages, respectively, which may potentially modulate leukocyte influx to the adventitia. This ligand-receptor pair analysis predicted the manner in which resident mesenchyme cells interacted with and attracted immune cells in vivo.
CONCLUSION
By providing transcriptomic profiles at single cell resolution, scRNAseq is a powerful tool to study heterogeneity and dynamic changes in cell populations.93738394041 However, scRNAseq has limitations, such as biases in transcript coverage and low capture efficiency.4243 We discussed how to handle some of these limitations in the pre-processing section.
In this review, we discussed certain scRNAseq studies about atherosclerosis which tried to identify heterogeneous cell populations by scRNAseq assay. Focusing on the data analysis aspect, we discussed some of the computational approaches that may be applied to the study of atherosclerosis. Clustering has been applied to all studies and has contributed to the identification of previously uncharacterized populations.111213151617 Additionally, clustering various computational algorithms has provided new angles in studying atherosclerosis. For instance, pseudo-time analysis revealed key genes associated with atherosclerosis development.16 Cell-to-cell communications was investigated by studying the co-expression of ligand-receptor pairs.14 We also discussed spatial transcriptomic analysis which attempts to provide information about the locations of cells in the cell transcriptome. This can be used to identify tissue-morphology-specific gene expression.
There have been attempts to provide additional data collected from the same set of cells. G&T-seq44 provides information on DNA methylation as well as transcriptome from the same set of cells at the same time. TARGET-seq detects genetic mutations which together with transcriptome from a single cell.45 However, TARGET-seq only detects a limited number of known mutations. In comparison, SIDR46 enables parallel sequencing of the entire genomic DNA and messenger RNA of a cell. CITE-seq47 and REAP-seq48 quantify transcriptome with protein level using antibodies conjugated to a tripartite DNA sequence that contains a primer for amplification and sequencing (polymerase chain reaction handle), a unique oligonucleotide that acts as an antibody barcode, and an oligo (dA). These technologies can provide new insights into scRNAseq analysis. Overall, single-cell-resolution data will enhance our understanding about the development of morbid metabolic disorders such as atherosclerosis.
 XML Download
XML Download