

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cerebrovascular disease (CVD) and Alzheimer's disease (AD) are common causes of dementia.1 The pathological characteristics of CVD include structural and functional changes in the vasculature that affect oxygen and nutrient delivery to the brain.2 Several risk factors contribute to the development of CVD, including diabetes, obesity, hypertension and smoking.3 CVD and cognitive decline have been increasingly recognized as diabetic complications.45 Despite recent advances in the care of patients with Type 2 diabetes (T2D), the risk of CVD and dementia remains higher (up to 2-fold) in the population with T2D or prediabetes compared to metabolically normal individuals.67 The causative mechanisms underlying diabetes-associated cognitive dysfunction and dementia remain elusive. Clinical evidence indicates that overt hyperglycemia is linked to diabetes-associated cognitive decline8 and dementia risk in non-diabetic individuals.9 Multi-centered, randomized controlled studies have shown, however, that stringent control of glycemic parameters, i.e., glycated hemoglobin (hemoglobin A1c) and fasting plasma glucose levels, does not improve cognitive function,1011 rather, it increased mortality in patients with dementia.11 On the other hand, repeated hypoglycemic episodes are clearly linked to cognitive decline and increased dementia risk.812 From these studies, we foresee a need to identify novel contributing factors to the risk of cognitive impairment in humans with T2D or prediabetes.
Amylin, also known as islet amyloid polypeptide (IAPP), is a hormone co-synthesized with insulin by pancreatic β-cells and participates in the central regulation of satiety.13 Individuals with prediabetic insulin resistance have hypersecretion of both insulin and amylin.13 Thus, hyperamylinemia coincides with hyperinsulinemia and prediabetic insulin resistance, always.
Recent studies show that amylin accumulates in the cerebral small vessels of patients with dementia and T2D.1415 Amylin deposition and mixed amylin-β amyloid (Aβ) plaques were also detected in brains of T2D patients with pathological AD.1516 These results141516 suggest a pathological role of amylin dyshomeostasis in the development and progression of CVD and dementia. Here, we review clinical evidence of amylin pathology in CVD and dementia, discuss currently available animal models for diabetes and dementia, identify existent challenges and suggest future work that could lead to uncovering novel molecular mechanisms underlying the impact of diabetes on brain function.
PERIPHERALLY-MEDIATED AMYLIN DYSHOMEOSTASIS PROVOKES CEREBRAL SMALL VESSEL DISEASE
Amylin is synthesized and co-secreted with insulin by the pancreatic β-cell.13 The amylin peptide crosses normally the blood-brain barrier (BBB)17 and binds to neurons in the cerebral feeding centers to regulate satiety.1819
Amylin from several mammalian species, including humans, cats, dogs, and monkeys (but not rodents), forms amyloid when overexpressed.13 Most of the patients with T2D (>90%) have amylin amyloid deposition in the pancreatic islets.1320212223 Aggregated amylin induces β-cell dysfunction and apoptosis, contributing to the gradual loss of β-cell mass.23 Recent studies report that patients with T2D have abnormal accumulation of aggregated amylin in extra-pancreatic tissues, including the kidneys24 and the heart.25 Accumulating evidence indicates that the presence of amylin deposition in brains of patients with T2D and dementia.14151626272829 Amylin accumulation is particularly abundant in the cerebral small vessels.1415 We showed that microvascular amylin accumulation is associated with infarction and perivascular astrocyte recruitment, indicative of microvascular injury.14 Schultz et al.27 showed that amylin forms intracellular inclusions in the brain microvascular pericytes of AD patients with T2D. Amylin-containing pericytes showed fragmented nuclei and loss of neuron-glial antigen 2 expression, which is necessary for pericyte viability and function.27 Additionally, amylin appears to modulate pericyte autophagy and induces higher toxicity compared to Aβ amyloid in vitro.27 Results from the same research team showed the presence of aggregated amylin in the retinal microvessels.29 Capillary amylin accumulation in the retina appears to correlate with hippocampal amylin angiopathy and amylin burden.29 These results141516272829 suggest the possibility of using peripherally-mediated amylin dyshomeostasis as a biomarker of cerebrovascular risk in patients with T2D.
AMYLIN DYSHOMEOSTASIS AS A CONTRIBUTING FACTOR TO NEURODEGENERATION
One potential mechanism underlying vascular contributions to cognitive impairment and dementia (VCID) involves the interaction of diabetes-related amylin dyshomeostasis with AD pathology (Fig. 1). A study by Westermark et al.13 shows that the level of amylin is increased in the diffuse and dense cerebral plaques and within vascular amyloid deposits in brains of patients with pathological AD, without the clinical diagnosis of T2D.16 The results are consistent with previous data showing that ex vivo cross-seeding by amylin aggregates exponentially promote mixed amylin-Aβ amyloid formation.16 In line with these findings, a recent study demonstrates that mice expressing a mutated form of the amyloid precursor protein (APP) in neurons and human amylin in the pancreatic islets develop mixed amylin-Aβ plaques in the brain,30 similar to the pathology observed in individuals with T2D and AD.1516 When compared to transgenic mice expressing only the human amylin or the mutated form of APP protein alone, mice with mixed amylin-Aβ pathology show accelerated neurological dysfunction.30 The mechanism for exacerbated neurological function in this mice model is postulated due to the increased Aβ burden in association with elevated amylin accumulation in the brain.30 In contrast, the clinical data shows that Aβ burden is not increased in patients with T2D and AD.3132 Thus, further investigations will be necessary to clarify the specific mechanism(s) underlying the effects of amylin dyshomeostasis on the brain in humans.
Fig. 1
Diabetes-related amylin dyshomeostasis leads to the formation of pancreatic amyloid and promotes amylin accumulation in the peripheral circulation. Circulating oligomerized amylin deposits in the brain microvasculature and induces small vessels injury by deranging the microvascular endothelium. Oligomerized amylin also forms mixed amylin-Aβ plaques in the brain parenchyma, accentuating neurotoxicity. Diabetes-related amylin dyshomeostasis promotes a feed-forward pathological process by which circulating oligomerized amylin injures brain microvasculature and synergizes with Aβ pathology to induce dementia.
Aβ, β amyloid.
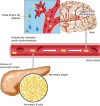
The increased propensity of amylin and Aβ to interact may be due to the fact that the amino acid sequence of human amylin is 52% similar to that of Aβ1-40 and Aβ1-42. The studies mentioned in above demonstrate the Aβ-seeding ability of human amylin in the brain. Nonetheless, one cannot exclude that the Aβ-seeding for amylin deposition is also a possible process that may occur parallel or independent from amylin-seeding Aβ.
In addition to protein-protein interaction, intriguingly, amylin and Aβ interaction are also found at the genetic level. Genome-wide association analysis further supports the interaction between the amylin gene and AD pathophysiology.33 Specifically, near genome-wide significant interaction effect was observed for an imputed variant rs73069071, located on chromosome 12p 12.1, within the amylin gene.33 The rs73069071-by-Aβ deposition was found for interaction effect on global cognitive function in AD patients.33 Although remaining elusive, the postulated mechanism involves the effect of this single nucleotide polymorphism on amylin production, whose product is known to interact with Aβ or possibly altering Aβ metabolism, hence modulating the impact of Aβ-deposition on cognitive performance.
Within the effort to understand the interaction of amylin and Aβ pathology, results from several studies suggest that the interaction is mediated through amylin receptors.3435 Experimental data suggest that Aβ directly activates amylin receptors, and that the oligomerized Aβ-induced neurotoxicity can be blocked with specific amylin receptor antagonists or downregulation of amylin receptor expression.3435 Interestingly, amylin receptor expression in the brain was up-regulated within the area of increased amyloid burden in AD transgenic mice which overexpress the mutated form of APP.35 Thus, while amylin accumulation appears to have deleterious effects, overexpression of amylin receptor (by the unknown mechanisms) possibly provides a platform for Aβ-induced neurotoxicity.
Besides pathological interaction of amylin with Aβ, aggregated amylin itself is neurotoxic. Several mechanisms of amylin-induced neuronal toxicity, including calcium dysregulation, oxidative stress, and mitochondrial dysfunction have been proposed.283436 In neurons, amylin forms adduct with 4-hydroxynonenal, a marker of peroxidative membrane injury, leading to increased synthesis of the pro-inflammatory cytokine interleukin 1β.28 The amylin-mediated neuronal injuries were blocked ex vivo by membrane stabilizers and lipid peroxidation inhibitors.28 Thus, aggregated amylin deranges the lipid membrane and predisposes it to peroxidative injury and inflammatory responses.
Given the pathological function of aggregated amylin,141537 the interaction of amylin with Aβ may accentuate neurotoxicity. Therefore, prevention strategies to effectively block amylin-mediated neurodegeneration should involve the inhibition of amylin-induced neuronal membrane injury and amylin-Aβ interaction.
ANIMAL MODELS FOR CONTRIBUTIONS OF T2D TO COGNITIVE IMPAIRMENT AND DEMENTIA
Rodents are the most commonly used laboratory models for diabetes and dementia owing to their 99% similarity to the human genome.38 However, neither diabetes nor dementia-like pathology spontaneously occurs in rodents. Conditions associated with diabetes, dementia, or both can be induced in rodents.1437394041 For the most part, insights from these interventions have been restricted to cerebral effects of inducing diabetes in normal rodents and in rodents genetically modified to develop neurodegeneration linked to the accumulation of Aβ in the brain.
In humans with T2D, the variation in the clinical phenotype of cognitive dysfunction or dementia may result from multiple pathologies.3142 An existent challenge of using rodent models to uncover mechanisms underlying the impact of T2D on brain structure and function lies on the fact that some genetic modifications that lead to T2D phenotype directly impact brain structure and function. For example, leptin or leptin-receptor deficiency, which is the commonly used genetic modification to induce obesity-related metabolic disturbances in rodents,43 has negative effects on learning and memory.44 Thus, interpretations made on specific molecular mechanisms of diabetes-induced brain dysfunction need careful consideration.
Overexpression (3-fold) of human amylin in rats leads to the mid-life onset of hyperglycemia linked to pancreatic amylin amyloid and β-cell apoptosis.45 Diabetes in human amylin expressing rats (HIP rats) is associated with neurological deficits including declined learning and memory, vestibulomotor dysfunction, altered balance and gait abnormalities.1437 In brains of the HIP rats with motor abnormalities, amylin deposition in small blood vessels correlates with microhemorrhages, decreased tight junction proteins levels and perivascular astrocyte activation,14 indicating that systemic amylin dyshomeostasis damages the BBB. Magnetic resonance imaging of HIP rat brains shows increased ventricular volumes with white matter hyperintensities and brain atrophy.14 Further assessment of cerebral blood flow in HIP rats via intravenous infusion of fluorescent microspheres and the measurement of the level of fluorescent microspheres retrieved from the brain capillaries reveal that amylin dyshomeostasis is associated with capillary loss and decreased cerebral perfusion.14 Amylin dyshomeostasis occludes or/and damages the brain small vessels, leading to brain parenchymal loss. Therefore, the animal model with amylin dyshomeostasis has the advantage to recapitulate major lesions, vascular lesion and brain atrophy, seen in brains of individuals with T2D (; for a review31).
CURRENT CHALLENGES AND FUTURE DIRECTIONS FOR LABORATORY STUDIES
Investigations of human brain tissues and rodent models for T2D suggest that amylin dyshomeostasis could play a potential role in the development of CVD and AD in individuals with T2D. Because amylin is produced in the pancreas and accumulates in the brain in association with CVD and AD, it is of clinical interest to decipher the central aspects of amylin dyshomeostasis in diabetes-related CVD and dementia. First, cerebrovascular accumulation of amylin may require carrier(s) in the circulation. The possible carrier could be macro-/micro-molecules or cellular components in the circulation. The animal model for amylin dyshomeostasis discussed above serves as a useful tool for extensive in vivo characterization of pharmacological interventions. Second, the translocation of amylin from the blood vessels to the parenchyma may require protein transporter(s). Likely, such translocation is concentration-dependent, because high aggregated amylin concentration is found to directly induce endothelial apoptosis in our previous study.14 Following the assessment of the cerebrovascular injury in the animal model of amylin dyshomeostasis, in vitro studies designed to identify specific protein transporter(s) are an important step to decipher how amylin interacts with BBB units and its translocation mechanism(s). Third, the specific function of amylin and aggregated amylin in vascular, neuronal and glial cells have not been fully understood. From our previous studies, we found that aggregated amylin could directly participate in cerebrovascular injury14 and neurodegeneration.28 However, much less is known about the effects of aggregated amylin on vascular, neuronal and glial function. Finally, we propose that understanding the pathways for aggregated amylin clearance from the circulation and the brain vasculature could be beneficial for therapeutic development.
Indeed, future research should take into consideration that the pathophysiology of T2D involves a complex interaction of multiple deficiencies. Therefore, the multi-facets of the T2D requires research models that can represent the complexity of the disease, in order to understand the underlying mechanisms attributed to CVD or dementia.
CONCLUSION
Diabetes-related amylin dyshomeostasis has a pathological role in CVD and dementia. Existent evidence has suggested that the accumulation of aggregated amylin in the brain blood vessels and brain parenchyma, mediated by peripheral amylin dyshomeostasis, is a potential mechanism underlying VCID.
 XML Download
XML Download