

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Creutzfeldt-Jakob disease (CJD) is a fatal neurodegenerative disease characterized by rapidly progressive dementia, cerebellar ataxia, myoclonus, parkinsonism, or various neurobehavioral symptoms due to tissue deposition of a misfolded prion protein.1 A genetic variant in the prion protein gene (PRNP) is the most common cause of familial CJD (fCJD) which accounts for only 10%–15% of all CJD patients.23 We report a rare case with a nucleotide substitution of an isoleucine for a valine at codon 180 (V180I) in PRNP, who presented with visual hallucinations and illusions.
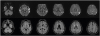
An age 78 male without comorbidity visited the hospital because of visual symptoms. He complained of visual hallucinations (e.g. strangers were shown) and visual illusions (e.g. a chair appears as a child) that began five months ago and has been aggravated at night. He became anxious about the symptoms. His family reported cognitive decline in the last three to four months. At admission, the physical and neurological examinations were unremarkable except for the visual symptoms. He showed neither ataxia nor pyramidal/extrapyramidal tract signs. On detailed neuropsychological tests, the Mini-Mental State Examination score was 23 and the clinical dementia rating score was 1. He showed cognitive impairment in multiple domains including attention, naming, verbal and visual memory, visuospatial, and frontal executive functions. On brain magnetic resonance imaging (MRI), bilateral high signal intensities including frontal, parietal, temporal, and occipital cortex were shown in axial diffusion weighted and fluid-attenuated inversion recovery images. The caudate nucleus, putamen, thalamus, and cerebellum were spared (Fig. 1). Cerebrospinal fluid (CSF) examination showed no white blood cells, normal glucose, and mildly elevated protein (64.5 mg/dL). CSF evaluations including infection markers were all negative. An electroencephalography (EEG) was normal. After discharge, fCJD was confirmed by that the 14-3-3 protein in CSF was weakly positive and sequencing of PRNP revealed a V180I mutation from GTC (valine) to ATC (isoleucine) with methionin homozygosity at codon 129 (129 M/M) and glutamate homozygosity at codon 219 (219 E/E) (Fig. 2). CSF total tau level prominently increased (1,956.0 pg/mL; normal range<200 pg/mL) and Aβ42 protein level moderately decreased (101.6 pg/mL). CSF pathogenic prion protein (PrPSc) using real-time quaking-induced prion conversion assay was negative. He has been taking symptomatic medications and undergoing follow-up evaluations for three months with no clinical progression or improvement.
Our patient revealed progressive dementia and visual symptoms during five months without other clinical symptoms such as ataxia, parkinsonism, myoclonus, or pyramidal tract signs. This phenotype resembles a Heidenhain variant of CJD, a peculiar clinical presentation of sporadic CJD characterized by visual disturbances at disease onset and representing early involvement of prions to the occipital cortex.4 Intriguingly, considering that visual disturbance is rare in this type of fCJD,3 our case showed atypical presentations including visual hallucination and illusions. A few fCJD cases with V180I mutations were previously reported in South Korea. However, presenting symptoms, duration, and neuroimaging findings varied. Two cases presented with subacute progressive dementia and neuropsychiatric symptoms35 and the other 2 cases with sudden stroke-like symptoms such as mental change or weakness.67 The previous cases were different from our case in that they showed multiple neurological abnormalities including ataxia, pyramidal/extrapyramidal tract signs, aphasia, neuropsychiatric symptoms, and akinetic mutism.3567
In regards to CSF markers, elevated levels of protein 14-3-3 and/or t-tau, which reflect neuronal damage, are recommended by diagnostic criteria for CJD although amyloid β (Aβ) levels shows conflicting results.3 In our patient, CSF tau protein prominently increased, more than that in Alzheimer's disease (AD), suggesting CJD than AD pathologies. Reduced Aβ42 levels have also been found in CJD in the absence of cortical amyloid plaque.3 In regards to PrPSc, fCJDs are known to show fewer positives (approximately 65%–70%) when compared with those in sporadic cases (approximately 90%).3 Because V180I fCJD are characterized by later onset (approximately age 75 or older), low penetration rate, and may show only cognitive decline, it can be misdiagnosed as AD.3
In summary, V180I fCJD patient may show relatively slow clinical progression without signs such as myoclonus, ataxia, or pyramidal/ extrapyramidal signs. They also may show negative findings in EEG or PrPSc, increased tau and decreased Aβ42 levels in CSF, hence mimic AD. The only sign for differential diagnosis could be diffuse bilateral hyperintensities using diffusion MRIs. Thus, we should consider fCJD, even though the patient has no family history, and conduct genetic evaluations for fCJD in patients with progressive visual disturbance and dementia.

 XML Download
XML Download