

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome (MIM 208085) is a rare autosomal recessive disorder first reported in 1973 by Lutz-Richter and Landolt in the offspring of a marriage between close relatives [1]. Its characteristic clinical features are arthrogryposis, renal tubular acidosis, and neonatal cholestatic jaundice. ARC syndrome is sometimes associated with additional presentations, including ichthyosis, agenesis of the corpus callosum, congenital cardiovascular abnormalities, nephrogenic diabetes insipidus, hypothyroidism, recurrent sepsis, deafness, and platelet abnormality [2]. The locus that causes this disorder is located on chromosome 15q26.1, and germline mutations were identified in vacuolar protein sorting 33 homolog B (VPS33B) and VPS33B-interacting protein, apical-basolateral polarity regulator (VIPAR) genes [3]. ARC syndrome is lethal, with death generally occurring in the first year of life. Mild, atypical symptoms at birth and in the first few weeks after birth result in delayed treatment of this disorder. In this case study, we describe an infant who was diagnosed with ARC using targeted exome sequencing (TES) before symptoms became apparent.
The Institutional Review Board (IRB) of Korea University Guro Hospital approved this study (No. 2019GR0272). Written consent was waived under the IRB's approval.
CASE REPORT
A female neonate was born at the gestational age of 41 weeks and 2 days via cesarean section because of progression failure. Her birth weight was 3.36 kg and the parents were not consanguineous. There was no family history of hepatobiliary disease. She had a coarse face, a high arched palate, ichthyosis, hepatosplenomegaly, and arthrogryposis with bilateral dislocation of the hips, flexion contracture of the knee joints, and a vertical talus (Fig. 1). She suffered from generalized hypotonia and respiratory distress, and a nasogastric tube (NG) was placed immediately after birth because of impaired sucking and swallowing.
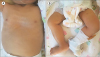 | Fig. 1The dysmorphic features associated with arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. An infant with ARC syndrome presenting symptoms of (A) ichthyosis and (B) arthrogryposis manifested by bilaterally dislocated hips, flexion contracture of the knees, and vertical talus.
|
At birth, the patient's hemoglobin level was 14.9 g/dL, her leucocyte level was 2,900/μL, and her platelet level was 157,000/μL (normal platelet morphology). Her initial electrolyte profile was normal. Levels of serum total bilirubin and direct bilirubin were 1.71 mg/dL and 0.55 mg/dL, respectively. At 7 day after birth, laboratory evaluation revealed cholestasis (total bilirubin 15.91 mg/dL, direct bilirubin 4.05 mg/dL) with a normal gamma glutamyl aminotransferase (GGT) level (32 IU/L). Serum protein and albumin levels were 5.1 and 3.3 g/dL, respectively. Prothrombin and partial thromboplastin times were normal. At 12 d after birth, the total bilirubin level had increased to 25.94 mg/dL. Hepatitis viral markers and neonatal metabolic screening test results were negative.
Magnetic resonance cholangiopancreatography (MRCP) revealed poor delineation of the biliary tree. Liver biopsy revealed paucity of bile ducts, giant cell transformation, and some lipofuscin pigments, suggestive of biliary atresia (BA) or progressive familial intrahepatic cholestasis (PFIC) (Fig. 2). Regardless of the MRCP and biopsy findings, genetic studies were conducted for dysmorphism and normal GGT levels to exclude disorders such as ARC syndrome. TES was performed using peripheral blood samples from the parents and the patient after obtaining written informed consent. All sequences were BLAST searched against the TruSight One Sequencing Panel (https://support.illumina.com/sequencing/sequencing_kits/trusight_one_kit.html) to determine variations, and single nucleotide polymorphisms were excluded using the Genome Aggregation Database [4], Exome Aggregation Consortium [5], 1,000 Genome Project [6], and Korean Reference Genome Database (http://coda.nih.go.kr/coda/KRGDB/index.jsp). Exome sequencing together with Sanger sequencing revealed compound heterozygous mutations, c.707A>T and c.239+5G>A, in VPS33B. c.707A>T, inherited from the father, is a novel variant. c.239+5G>A, inherited from the mother, is a rare variant found in less than 0.1% of the general population (Fig. 3).
 | Fig. 2Micrographs from the liver biopsy of arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome patient. Micrographs from the histological examination (liver biopsy) of patient with ARC syndrome showing (A) giant cell hepatitis, (B) intrahepatic hematopoiesis, (C) lipofuscin deposition, and (D, E) a paucity of bile ducts (A-D: Sections were stained with hematoxylin and eosin and viewed under 400×magnification; E: Sections were stained with CK19 and viewed under 200×magnification).
|
 | Fig. 3Missense mutations found in a neonate with cholestasis. Molecular analysis of a neonate with cholestasis revealed compound heterozygous mutations in vacuolar protein sorting 33 homolog B. The DNA chromatograms highlighting the missense mutations: (A) c.707A>T (p.Asp236Val) and (B) c.239+5G>A. The c.707A>T mutation was a novel variant that was acquired from the father.
|
At 15 day, urine analysis indicated proteinuria, global aminoaciduria, glucosuria, hypercalciuria, hyperuricosuria, and hyperphosphaturia with metabolic acidosis (normal anion gap), suggesting renal tubular acidosis. There was hydronephrosis of both kidneys, but no nephrocalcinosis was observed on abdominal ultrasonography. Brain magnetic resonance imaging suggested agenesis of the corpus callosum. Thyroid function evaluation indicated levels of free thyroxine at 1.15 ng/dL and thyroid stimulating hormone levels greater than 100 μIU/mL. Congenital hypothyroidism with an ectopic thyroid gland was diagnosed. Neonatal hearing screening revealed sensorineural hearing loss on both sides.
She was discharged from the neonatal intensive care unit at 82 days of age but was readmitted frequently because of recurrent infections. At 6 months of age, growth was still faltering despite adequate calorie intake via NG tubes. While her cholestasis improved slightly, her renal function was impaired because of severe renal Fanconi syndrome with signs of nephrogenic diabetes insipidus. Despite continuous renal replacement therapy, she died at the age of 9 months from renal failure secondary to sepsis.
DISCUSSION
Our patient exhibited all the classical clinical features of ARC syndrome, with comorbid presentations of ichthyosis, agenesis of corpus callosum, deafness, hypothyroidism, and nephrogenic diabetes insipidus. Previous cases suggested a usual pattern of liver histology including paucity of bile ducts, lipofuscin deposition, and giant cell hepatitis [1789101112]. Eastham et al. [13] argued that absence of such changes might be unimportant, because histopathology depends on the timing and site of biopsy. The pathology of cholestasis is nonspecific; therefore, pathologists adhere to rigorous criteria to avoid over interpretation of histological findings. Additional diagnoses (i.e., TES) are required before pathogenesis can be confirmed. Furthermore, ARC syndrome patients might have platelet abnormalities and are vulnerable to coagulation. Thus, liver biopsy can lead to the risk of lethal bleeding.
In differential diagnosis, BA, which is first considered in neonates with cholestasis, may be excluded by the presence of normal bile ducts on diagnostic images and normal serum GT levels [14]. Similar clinical and laboratory results might be evident in ARC syndrome, PFIC, and bile acid synthesis disorders (BASDs). PFIC is an autosomal recessive disorder of cholestasis, which causes cholestasis and hepatocellular damage resulting from bile acid transport defects. BASDs are a group of metabolic disorders characterized by defects in the production of normal bile acids, and the accumulation of unusual bile acids and intermediary metabolites. If clinical symptoms are not fully manifested in the patient, additional genetic testing should be considered to distinguish ARC syndrome from other diseases [15].
Family history, classical clinical presentations, and genetic mutations should be evaluated for accurate diagnosis and early initiation of tailored treatment [14]. In our patient, invasive techniques, such as liver biopsy, did not contribute to a differential diagnosis of ARC syndrome, whereas early TES, together with the clinical presentations, constituted an apparently accurate diagnostic procedure.
ARC syndrome was mapped to chromosome 15q26.1 and germline mutations in VPS33B were identified by Gissen et al. [3] in 14 kindreds with ARC syndrome. VIPAR is another causative gene of ARC syndrome. VPS33B is crucial in intracellular vesicular trafficking pathways, while VIPAR exerts pleiotropic effects on polarity and apical membrane protein restriction via the formation of VPS33B-VIPAR complexes, ensuring a normal cellular structure. These proteins are found in many parts of the body, including the kidneys, liver, heart, lungs, brain, skin, and skeletal muscles, accounting for the multisystemic symptoms characteristic of the ARC clinical phenotype [16171819].
To allow researchers easy access to updated information on global genetic epidemiology, the online, locus-specific Leiden Open-Source Variation Database for ARC syndrome was created in 2011. This database includes a total of 228 unique variants in VPS33B and 34 unique variants in VIPAR, of which sequence mutations are categorized as ‘pathogenic,’ ‘probably pathogenic,’ ‘no known pathogenicity,’ ‘probably no pathogenicity,’ and ‘effect unknown,’ depending on their predicted effects on the protein and the clinical phenotype [20]. In our case, exome sequencing revealed compound heterozygous mutations of c.707A>T and c.239+5G>A in VPS33B.
Notably, c.707A>T is a novel variant that has not been reported among the 123,136 variants in the general population. According to the PolyPhen-2 program, which predicts protein functional defects, a c.707A>T score of ‘1.000’ was ‘probably damaging.’ Scores obtained using SIFT, FATHMM, LRT, MutationTaster, MutationAssessor, PROVEAN, and VEST3 (http://asia.ensembl.org/info/genome/variation/index.html) via in silico analyses were ‘0.000,’ ‘−4.030,’ ‘0.000,’ ‘1.000,’ ‘3.560,’ ‘−8.600,’ and ‘0.988,’ respectively, indicating that this mutation mighty cause functional damage.
Furthermore, c.239+5G>A in VPS33B is rare, being found in less than 0.1% of the general population. Adaptive boosting and random forest scores from dbscSNV19 (http://asia.ensembl.org/info/genome/variation/index.html) were ‘1.000’ and ‘0.996,’ respectively, suggesting that the mutation was pathogenic and affected splicing. Functional analyses should be conducted to determine the effect of this missense mutation on protein function and patient phenotypes.
In this study, we reported a patient with ARC carrying a novel VPS33B mutation, determined using TES. Liver biopsy was non-specific and did not contribute to the differential diagnosis. Early TES, together with clinical presentations, constituted an apparently accurate diagnostic procedure. No specific treatment currently exists for ARC, therefore, early recognition and genetic diagnosis is essential to predict and prepare for the course of this disease.
 XML Download
XML Download