

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Gastric cancer (GC) is one of the most common malignancies as well as the leading cause of cancer mortality worldwide, with more than 1,000,000 new cases (5th) and 783,000 deaths (3rd) reported in 2018 [1]. Korea has the highest incidence rate, with 25,800 new cases and 7,100 deaths expected in 2018 [2]; since a nation-wide screening program began in 1999, the number of early (E)GC cases has increased [34]. EGC treatment involves minimally invasive surgery including laparoscopic and robotic surgery, and endoscopic resection. The prognosis after surgery without additional treatment is excellent, with a 5-year overall survival (OS) rate over 90% [567891011]. However, a significant number of patients are diagnosed at an advanced stage, which is associated with poor outcome. Over 50% of patients with locally advanced GC experience tumor recurrence in their lifetime, even after radical surgery and adjuvant chemotherapy [12].
GC is a heterogeneous disease with various treatment outcomes. Patients with locally advanced GC receive postoperative chemotherapy after curative surgery in order to eliminate any chances of micrometastasis [1314]. However, this benefits only a limited number of patients, whereas others are overtreated or inappropriately treated. For example, in the CLASSIC trial [15], over 50% of patients who did not undergo adjuvant chemotherapy did not exhibit relapse and around 70% were still alive after 5 years. On the other hand, around 30% of patients had relapsed at 5 years and 20% had passed away even after cytotoxic chemotherapy. This data suggests that patient stratification based on the biological characteristics of the tumor, beyond the anatomical extent of the tumor (tumor-node-metastasis [TNM] staging), is needed.
GC is classified according to histomorphologic features. The Lauren classification categorizes GC into intestinal, diffuse, and mixed types, while the World Health Organization divides GC into papillary, tubular, mucinous, and poorly cohesive carcinomas [1617]. Despite its versatility and wide application in the clinic, a histomorphologic classification system, unfortunately, has limited utility in GC owing to its weak association with clinical parameters such as prognosis or responsiveness to chemotherapy.
Motivated by the lack of clinically relevant classification systems, the latest research on GC has focused on elucidating the molecular landscape including the genetics of this disease. Recent technological advances have enabled detailed investigation of the molecular characteristics of GC. Several GC classifications have been proposed based on the correlation between molecular profiles and clinical behavior [1819202122]. In addition, simplified patient classifiers have been suggested that can potentially help in determining the treatment approach, and novel preclinical models that recapitulate cancer in situ such as tumor organoids and patient-derived xenografts (PDXs) can bridge the gap between cancer genetics and tumor phenotype. This review discusses the molecular classifications of GC, simplified algorithms based on these classifications, and patient-derived models that can provide the next level of knowledge for implementing precision clinical care strategies for GC.
Go to : 
MOLECULAR CLASSIFICATION OF GC
The Cancer Genome Atlas (TCGA)
The TCGA Research Network Group [19] classifies GC into 4 molecular subtypes: Epstein-Barr virus (EBV), microsatellite instability (MSI), chromosomal instability (CIN), and genomically stable (GS). An analysis of 295 GC tissue samples using 6 molecular platforms,— i.e., array-based somatic copy number analysis, whole-exome sequencing, array-based DNA methylation profiling, messenger RNA (mRNA) sequencing, microRNA sequencing, and reverse phase protein array— revealed that EBV-associated GC accounted for 9% of the samples and had the highest rate of DNA hypermethylation. All EBV GC cases presented CDKN2A promoter hypermethylation and showed a clear difference from the MLH1 hypermethylation pattern. EBV subtype was strongly associated with mutations in PIK3CA (80%), ARID1A (55%), and B cell lymphoma 6 co-repressor (23%). Furthermore, EBV GC shows PD-L1/2 and JAK2 amplification and upregulation of immune cell signaling pathways. The EBV type is mainly observed in males and is typically located in the gastric fundus and body.
MSI tumors account for 22% of the total analyzed cases and exhibit hypermethylation of promoters, such as the MLH1 promoter, as well as high rates of mutation in PIK3CA (42%), KRAS/NRAS (25%), JAK2 (11%), human ERBB3 (14%), ERBB2 (5%), and EGFR (5%). Alterations in B2M and human leukocyte antigen-B that are 2 major histocompatibility complex class I genes, are commonly found in MSI GC, which is also associated with older age at diagnosis and female sex.
CIN occurs in 50% of all the GC cases. The CIN subtype is correlated with intestinal-type histology and aneuploidy and shows aberrant activation of receptor tyrosine kinase (RTK)/RAS signaling pathways, focal RTK amplification, and the highest frequency of TP 53 mutation (71%) as well as alterations in other canonical tumor suppressor genes such as SMAD 4 and APC. CIN tumors are frequently located at the gastroesophageal junction and gastric cardia.
The GS subtype constitutes 20% of GCs and exhibits diffuse histology. Mutations in CDH1 (37%) and RhoA (15%) and CLDN18/ GAP gene fusion (15%) are often detected in GS tumors, which also show upregulation of cell adhesion and angiogenesis-related pathways. Additionally, patients with the GS subtype are generally younger at the time of diagnosis.
TCGA has proposed a comprehensive set of molecular characteristics that define GC. Nevertheless, it failed to establish prognostic differences between each subtype. The TCGA cohort includes samples treated by different strategies, with only 47.7% of samples analyzed due to poor sample quality. Therefore, the clinical significance of TCGA subtypes had remained unclear. However, the clinical relevance of the TCGA subtypes was reconsidered in a recent study investigating the associations among survival, the benefits of chemotherapy, and TCGA subtypes in 2 independent cohorts [23]. The EBV and GS subtypes showed the best and worst prognoses, respectively, while the patients with the CIN and GS subtypes benefitted the most and least from adjuvant chemotherapy, respectively. Moreover, TCGA risk score based on subtype probability was found to be an independent prognostic factor in a multivariate analysis and showed a linear correlation with 5-year recurrence rate.
Asian Cancer Research Group (ACRG)
Based on an analysis of mRNA expression level, somatic copy number, and targeted gene sequencing in 300 tumors, the ACRG proposed the following 4 GC molecular subtypes: MSI (22.7%), microsatellite stable with epithelial-to-mesenchymal transition features (MSS/EMT, 15.3%), MSS with tumor suppressor p53 (TP53) activity (MSS/TP53, 26.3%), and MSS with TP53 functional loss (MSS/TP53−, 35.7%) [20].
MSI exhibited hypermutation, with mutations in KRAS, ALK, ARID1A, and PI3K pathway genes. The MSS/EMT subtype was characterized by loss of CDH1 expression and had the lowest number of mutation events among the 4 subtypes. MSS/TP53+ GC is often associated with EBV infection and is enriched in mutations in APC, ARID1A, KRAS, PIK3CA, and SMAD4 compared to the MSS/TP53− subtype. On the other hand, the latter has the highest frequency of TP53 mutation (60%) and focal amplification of HER2, EGFR, cyclin E1 (CCNE1), CCND1, MDM2, Robo2, GATA6, and MYC.
ACRG also characterized the clinical behavior of each GC subtype. MSI showed the best prognosis and lowest rate of recurrence, which was mainly hepatic. MSS/TP53+ had the next best prognosis, followed by MSS/TP53−. The MSS/EMT subtype had the worst prognosis and highest rate of recurrence, which was mostly peritoneal. MSI tumors predominantly occur in the antrum; over 60% of cases are of the Lauren intestinal type and are usually diagnosed at early stages (I–II). On the other hand, MSS/EMT GC exhibits diffuse histology and a large subset is signet ring cell carcinomas; this subtype is diagnosed at the advanced stages (III–IV) and at a younger age.
Singapore–Duke classification
The Singapore–Duke study analyzed the gene expression patterns in 248 gastric tumors and identified 3 subtypes (proliferative, metabolic, and mesenchymal) [21]. Proliferative GCs were characterized by elevated expression of cell cycle-related genes and frequent TP53 mutation, copy number amplification, DNA hypomethylation, and a Lauren intestinal type. Oncogenic pathways such as E2F, MYC, and RAS signaling were upregulated in this subtype. Metabolic GCs showed upregulation of metabolic and digestion-related genes that are expressed in normal gastric mucosa. This subtype also showed hyperactivation of the spasmolytic-polypeptide-expressing metaplasia pathway, which is related to gastric metaplasia. The mesenchymal subtype was characterized by increased expression of genes related to the cell adhesion, extracellular matrix (ECM)–receptor interaction, and focal adhesion and activation of EMT and cancer stem cell pathways. The mesenchymal subtype was associated with a Lauren diffuse type and low copy number as well as alterations in p53, transforming growth factor β, vascular endothelial growth factor (VEGF), nuclear factor κ light chain enhancer of activated B cells (NF-κB), mechanistic target of rapamycin (mTOR), and sonic hedgehog (Shh) signaling pathways.
The Singapore–Duke study also compared drug responses among GC subtypes. Cell lines of the metabolic subtype were more sensitive to 5-fluorouracil (5-FU) than the others, and patients with metabolic GC showed greater responsiveness to 5-FU chemotherapy. Metabolic GC showed lower expression of thymidylate synthase and dihydropyrimidine dehydrogenase proteins, which are related to 5-FU resistance and this might be a possible explanation for the higher 5-FU sensitivity. On the other hand, cell lines of the mesenchymal subtype were particularly sensitive to PI3K/AKT/mTOR inhibitors, consistent with the high activation of the mTOR pathway.
Despite the differences in genomic alteration and drug response, the 3 subtypes showed no significant differences in cancer-specific and disease-free survival. Only proliferative GC patients showed shorter disease-free survival in the multivariate Cox proportional hazard regression analysis, although a correlation between higher TNM stage and worse prognosis was noted.
Go to :
SIMPLIFIED ALGORITHMS WITH CLINICAL APPLICABILITY
Newly introduced molecular classifications of GC have provided insight into the heterogeneous nature of GC (Table 1). Each subtype is characterized by specific gene mutations and alterations in the signaling pathways as well as prognosis and response to chemotherapy. Nonetheless, these classifications have limited clinical applicability because they require complex molecular analyses that are not feasible in clinical practice. Therefore, simplified classifying algorithms based on clinically available diagnostic tools have been proposed.
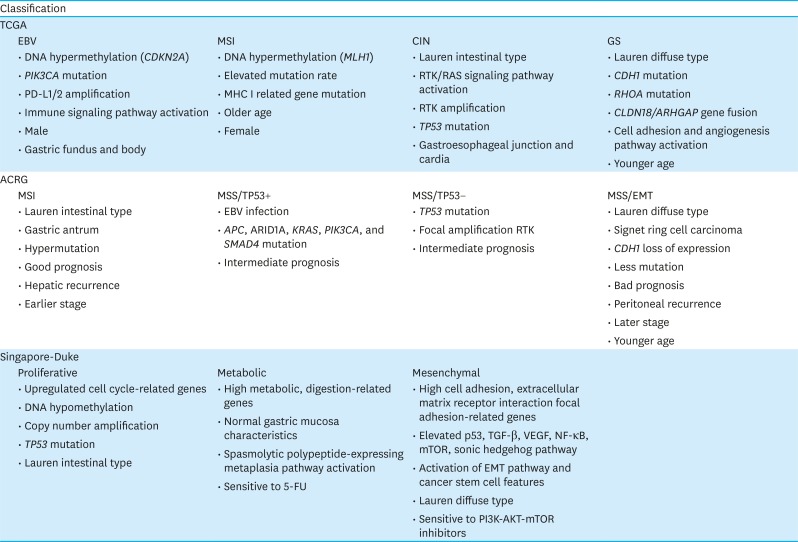
Table 1
Molecular classification of gastric cancer
TCGA = The Cancer Genome Atlas; EBV = Epstein-Barr virus; MSI = microsatellite instability; CIN = chromosomal instability; GS = genomically stable; PD-L1 = programmed death-ligand 1; MHC = major histocompatibility complex; RTK = receptor tyrosine kinase; ACRG = Asian Cancer Research Group; MSS = microsatellite stable; EMT = epithelial-mesenchymal transition; TGF = transforming growth factor; 5-FU = 5-fluorouracil; VEGF = vascular endothelial growth factor; NF-κB = nuclear factor κ light chain enhancer of activated B; mTOR = mechanistic target of rapamycin; TP53 = tumor suppressor p53.
![]()
Immunohistochemistry (IHC) and in situ hybridization (ISH)
The molecular spectrum of GC can be analyzed by tissue microarray analysis of protein and mRNA expression, allowing categorization of GC into subtypes with distinct characteristics (Table 2). A study of 438 GC patients treated with palliative chemotherapy using 10 GC panels, which included Epstein–Barr encoding region ISH, IHC for mismatch repair proteins, RTKs, phosphatase and tensin homolog (PTEN), and p53, revealed a relationship between protein/mRNA expression levels and the specific clinical characteristics of EBV-positive GC [24]. In an analysis of 244 gastric adenocarcinomas according to EBV positivity, MSI, TP53 and E-cadherin mutation, and Lauren classification revealed an association between IHC/ISH classification and clinicopathologic characteristics such as sex, tumor location, and OS [25].
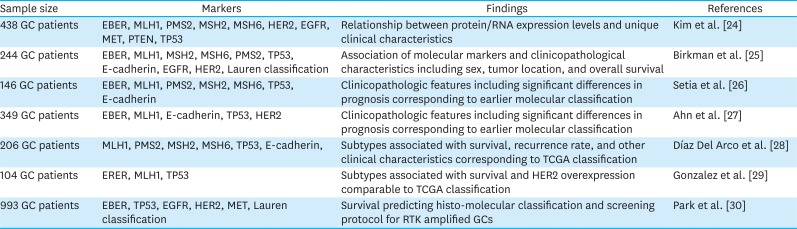
Table 2
GC classification algorithm based on immunohistochemistry and in-situ hybridization
| Sample size | Markers | Findings | References |
|---|---|---|---|
| 438 GC patients | EBER, MLH1, PMS2, MSH2, MSH6, HER2, EGFR, MET, PTEN, TP53 | Relationship between protein/RNA expression levels and unique clinical characteristics | Kim et al. [24] |
| 244 GC patients | EBER, MLH1, MSH2, MSH6, PMS2, TP53, E-cadherin, EGFR, HER2, Lauren classification | Association of molecular markers and clinicopathological characteristics including sex, tumor location, and overall survival | Birkman et al. [25] |
| 146 GC patients | EBER, MLH1, PMS2, MSH2, MSH6, TP53, E-cadherin | Clinicopathologic features including significant differences in prognosis corresponding to earlier molecular classification | Setia et al. [26] |
| 349 GC patients | EBER, MLH1, E-cadherin, TP53, HER2 | Clinicopathologic features including significant differences in prognosis corresponding to earlier molecular classification | Ahn et al. [27] |
| 206 GC patients | MLH1, PMS2, MSH2, MSH6, TP53, E-cadherin, | Subtypes associated with survival, recurrence rate, and other clinical characteristics corresponding to TCGA classification | Díaz Del Arco et al. [28] |
| 104 GC patients | ERER, MLH1, TP53 | Subtypes associated with survival and HER2 overexpression comparable to TCGA classification | Gonzalez et al. [29] |
| 993 GC patients | EBER, TP53, EGFR, HER2, MET, Lauren classification | Survival predicting histo-molecular classification and screening protocol for RTK amplified GCs | Park et al. [30] |
GC = gastric cancer; EBER = Epstein–Barr encoding region; MLH1 = mutL homolog 1; TP53 = tumor suppressor p53; PMS2 = PMS1 homolog 2; MET =mesenchymal epithelial transtion; PTEN = phosphatase and tensin homologue deleted on chromosome 10; HER2 = human epidermal growth factor receptor 2; EGFR = epidermal growth factor receptor; TCGA = The Cancer Genomic Atlas; RTK = receptor tyrosine kinase; MSH = mutS homologue.
![]()
A number of studies have proposed a GC classification algorithm based on the IHC and ISH results. Five subgroups of GC were defined using an algorithm integrating EBV positivity, MSI, and aberrant E-cadherin and p53 expression determined by IHC and ISH in American and Asian cohorts, respectively [2627]. These subgroups exhibited different clinicopathologic features including prognosis that were in accordance with a previously reported molecular classification. In a European cohort, 4 GC types were defined based on microsatellite stability and E-cadherin and p53 expression that showed differences in the survival, recurrence rate, and other clinical characteristics such as macroscopic morphology and Lauren classification [28]. Another study divided GC into 4 subgroups corresponding to TCGA classification based on EBV, MSI status, and p53 expression that differed in terms of survival probability and human epidermal growth factor receptor 2 (HER2) positivity [29]. A histo-molecular classification predicting patient survival based on Lauren classification, IHC, and silver-enhanced ISH has been established. These investigators also suggested a screening protocol for RTK-amplified GC, which has unique clinicopathologic features [30].
Single-patient classifier (SPC) based on 4-gene real-time polymerase chain reaction (RT-PCR)
An SPC predicting prognosis and response to adjuvant chemotherapy in patients with stage II–III resectable GC was developed [22]. The classifier is based on RT-PCR detection of the expression levels of 4 genes in formalin-fixed paraffin-embedded tumor tissue (FFPET). The 4 classifying genes—namely, GZMB, WARS, SFRP4, and CDX1—were identified from a transcriptome dataset consisting of 1,259 GC tumor specimens that were previously used in part to establish a molecular classification for GC. Unlike other molecular subtyping criteria for GC that are population-directed and heavily dependent on the composition of the dataset, and therefore not directly applicable to individual patients, the investigators first identified 5 molecularly distinct subclasses of GC with different clinical outcomes and selected 3 clinically actionable subtypes with specific gene modules (Table 3). They then evaluated the subtype-specific genes in various tissue types (fresh tissue vs. FFPET) using different assays (array or RNA sequencing vs. quantitative RT-PCR) to identify clinically useful biomarkers that can assign individual patients to a certain subtype. The patient classifier consists of a 2 rule-based, 2-tier classifying algorithm first characterizing the immune subtype and then the stem-like subtype for risk stratification, or the epithelial subtype for patient stratification based on the chemotherapy response. This approach was evaluated in a cohort of 307 patients and validated using an independent cohort consisting of patients (n=625) from the CLASSIC trial.

Table 3
GC classification by single patient classifier
EBV = Epstein-Barr virus; GC = gastric cancer; CTx = chemotherapy; MSS = microsatellite stable; MSI-L = microsatellite instability-low; TCGA = The Cancer Genome Atlas; GS =genomically stable.
![]()
The prognosticSPC divided patients into low-risk (immune), intermediate-risk (non-immune, non-stem-like), and high-risk (stem-like) groups, with 5-year OS rates of 83.2%, 74.8%, and 66%, respectively. The predictive SPC identified patients who will (epithelial type) from those who will not (immune or non-immune/non-epithelial type) benefit from chemotherapy. The former was responsive to adjuvant chemotherapy; the 5-year OS of these patients who underwent postoperative chemotherapy was higher than that of patients who had undergone only surgery (80%; 95% confidence interval [CI], 73.5–87.1 vs. 64.5%; 95% CI, 56.8–73.3). Whereas, no difference was observed between the treatment strategies in the latter. A subsequent study confirmed that the prognostic value of modified TNM staging based on the SPC was superior to that of conventional TNM staging [31].
Cheong et al. recommended applying their SPC for the stratification of the patients with localized resectable GC after surgery in order to facilitate the therapeutic decision-making [22]. Immune subtype patients may not need adjuvant chemotherapy after surgery as they generally have favorable prognosis, and additional chemotherapy does not improve their OS. On the other hand, it is recommended that the epithelial subtype patients undergo adjuvant chemotherapy after surgery, which seems to improve the survival in this group. These authors also highlighted the need for novel treatment options for the stem-like subtype, which has the worst prognosis and overlaps significantly with the chemotherapy no-benefit group [22].
Clinical implications of GC subtypes by SPC and relationship with other molecular subtyping schemes
The immune subtype in the SPC comprises of MSI and EBV, and largely overlaps with these TCGA subclasses as well as with the ACRG MSI subtype and with a part of MSS/TP53 subtype. The stem-like subtype largely overlaps with TCGA GS, ACRG MSS/EMT, and the Singapore–Duke mesenchymal subtypes, and has the poorest prognosis. The epithelial subtype overlaps with TCGA CIN, ACRG MSS/TP53−, and the Singapore–Duke metabolic subtypes. Notably, of the 3 representative molecular GC subtyping schemes preceding the SPC, only the Singapore–Duke study addressed the relationship between molecular subtype and response to standard chemotherapy. Only the metabolic subtype showed a correlation with 5-FU chemotherapy response. Given that the proliferative subtype shares molecular characteristics with TCGA CIN and that the latter benefits from chemotherapy, the findings of the Singapore–Duke group cannot be readily explained. The metabolic subtype also expresses mucin 5AC, which is a gastric protein that is also detected in some forms of intestinal metaplasia [22]. Intriguingly, the epithelial subtype described by Cheong et al., [22] which is the only molecular subtype whose predictive value has been demonstrated in randomized controlled CLASSIC trial samples, is characterized by high expression of CDX1, a marker for intestinal metaplasia. Although this unexpected association between the Singapore–Duke metabolic and SPC epithelial subtypes based on intestinal metaplasia warrants further investigation, it seems plausible that the biological properties related to intestinal metaplasia can enhance responsiveness to 5-FU-based standard chemotherapy in GC.
Go to :
PRECLINICAL MODELS IN GC
The paucity of useful preclinical models has impeded the development of targeted anti-cancer drugs [3233]. Cancer cell lines and cell line-derived xenograft mouse models have been used for preclinical studies in cancer research. However, these approaches cannot reproduce the biological complexity of cancer such as the tumor microenvironment and cancer cell heterogeneity. Moreover, most established cell lines are susceptible to genetic alterations owing to prolonged in vitro culture [3435]. Therefore, there is a need for novel preclinical models for GC [363738].
PDXs
PDXs are promising platforms for translational cancer research [32394041]. They are established by implanting patient tumor tissue into immune-compromised mice, and then directly transferring the resultant tumor from mouse to mouse. The implanted tumor fragments contain not only heterogeneous cancer cells but also part of the tumor microenvironment, thereby recapitulating the key characteristics of the parental tumor. PDX models are becoming an increasingly integral part of basic and translational research and are expected to improve the development of more effective novel therapeutics.
GC PDX models and key factors for successful engraftment have been reported by several groups. In one such study, 15 PDX models were established from surgical specimens of 62 GC patients that retained the histologic and genetic profiles of the original tumor over many passages [42]. A diffuse histologic type, low tumor cell percentage, and longer ex vivo and processing times were negatively correlated with success rate. Similarly, another group established 63 PDX models from 185 gastric adenocarcinoma biopsy specimens in which the pathological features, HER2 expression, somatic genetic alterations, and chemosensitivity were comparable to those of the primary tumors [43]. Chemotherapy prior to biopsy was associated with lower engraftment success rates. In 17 PDXs derived from 54 surgically resected GC samples, histomorphology and key protein expression were similar to those of the patients' cancer tissue. It was also found that the tumor growth and survival rate in the mice increased from generation to generation [44]. A high incidence of lymphomagenesis was observed in 25 GC PDX models from 126 tumor tissues, which was associated with inflammation of the primary tumor [45]. It was suggested that treating the mice with rituximab treatment for a short period can prevent lymphoma formation without impeding engraftment [46].
GC PDX models are useful for evaluating novel therapeutic strategies. For instance, they have been used to confirm the anti-cancer efficacy of agents targeting cMET, maternal embryonic leucine zipper kinase, fibroblast growth factor receptor 2, HER2, epidermal growth factor receptor, and other molecules [47484950] as well as the synergistic effects of combination therapy with 2 different drugs [5152535455]; and demonstrate that inhibiting the dual specificity phosphatase 6 could overcome cisplatin resistance [51]. PDX models can also be used to assess the efficacy of immunotherapy. Treatment with a combination of urelumab (anti-human cluster of differentiation [CD] 137) and nivolumab (anti-human programmed death protein 1) suppressed tumor growth in PDX models generated using patient peripheral blood mononuclear cells [56]. Engineered CLDN18.2-specific chimeric antigen receptor-expressing T cells infiltrated into tumor tissue and eliminated cancer cells in CLDN18.2-positive GC PDX models [57]. Moreover, PDX models can facilitate the development of diagnostic tools. For example, 64Cu-2-(p-thiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid-trastuzumab was used to non-invasively detect the HER2 expressed due to GC in a PDX model [58], and near-infrared fluorescence-based heptamethine carbocyanine dyes were shown to be useful for tumor-specific imaging [59]. Additionally, CD44 and aldehyde dehydrogenase were found to be highly specific biomarkers for detecting chemoresistant GC stem cells in 37 GC PDX models [60].
Patient-derived organoids (PDOs)
PDOs are relatively new concepts in translational cancer research. These 3-dimensional culture systems are established by embedding tumor cells into ECM proteins, followed by treatment with appropriate factors. PDOs generated from various cancer types retain the genetic and histologic features of the original tumor, including their microarchitecture and physiology [616263646566676869]. PDOs are relatively low-cost, have short doubling time, and are more suitable for genetic manipulation than PDX, thereby enabling large-scale drug screening and molecular analysis [70717273].
Recent studies have described the establishment of a GC organoids (GCOs) that can simulate the disease characteristics. Exome and transcriptome datasets derived from a PDO biobank comprising of 63 organoids from 32 patients showed that they retained morphology, protein expression, genetic features, and molecular subtypes of the original tumors. Moreover, the clinical efficacy of recently introduced drugs such as napabucasin, abemaciclib, and the ataxia telangiectasia and Rad3-related kinase inhibitor VE-822 was demonstrated using GCOs [74]. Another study investigated the niche factor independency of organoid lines, optimized the culture conditions, and revealed the genotype-phenotype relationships in 37 GC PDO lines exhibiting molecular and histological diversity. The authors also demonstrated the efficacy of Wnt-targeted therapy for a subset of GCs by testing porcupine inhibitor in a patient-derived GCO xenograft model [75]. Moreover, silencing C8orf76 was shown to suppress tumor growth in GCOs [76], and the Hedgehog/Gli inhibitor GANT-61 decreased programmed death-ligand 1 expression and cell proliferation in GCOs [77].
Numerous studies have highlighted the clinical utility of patient-derived GCOs. Organoids established from surgically resected GC specimens and treated with epirubicin, oxaliplatin, and 5-FU showed response comparable to that seen after the patients received chemotherapy [78]. GCOs derived from endoscopic biopsies maintained the genomic profile of the primary tumor including response to cytotoxic chemotherapy [79]. Finally, GCOs exhibiting variable morphology, mutation profile, and chemotherapy response express the same proteins as the original tumor, suggesting that these organoids can be used as viable preclinical models to test the efficacy of trastuzumab or palbociclib [80].
Clinical perspectives of patient-derived model systems in precision medicine
Patient-derived models are expected to contribute to the development of novel targeted drugs for personalized cancer treatment and bridge the gap between bench and bedside by reproducing the interpatient and intratumor heterogeneity of cancer (Fig. 1). PDOs and PDXs can be frozen and stored in tumor biobanks after cataloging their molecular phenotypes. Such biobanks that reflect the patient population can be used for large-scale high-throughput drug screening and identification of biomarkers for consecutive clinical trials [32648182838485]. Co-clinical trials—i.e., the clinical trials conducted on both patients and models derived from them—can further accelerate drug development [62738687] by facilitating molecular, cellular, and clinical analyses to validate clinical outcome data and investigating the response and resistance to therapeutics. In clinical settings, patient-based ‘avatar’ models established from resected tumor tissue or circulating tumor cells and representing individual patients will assist clinicians in optimizing individualized cancer treatment through drug screening and validation [327388899091].
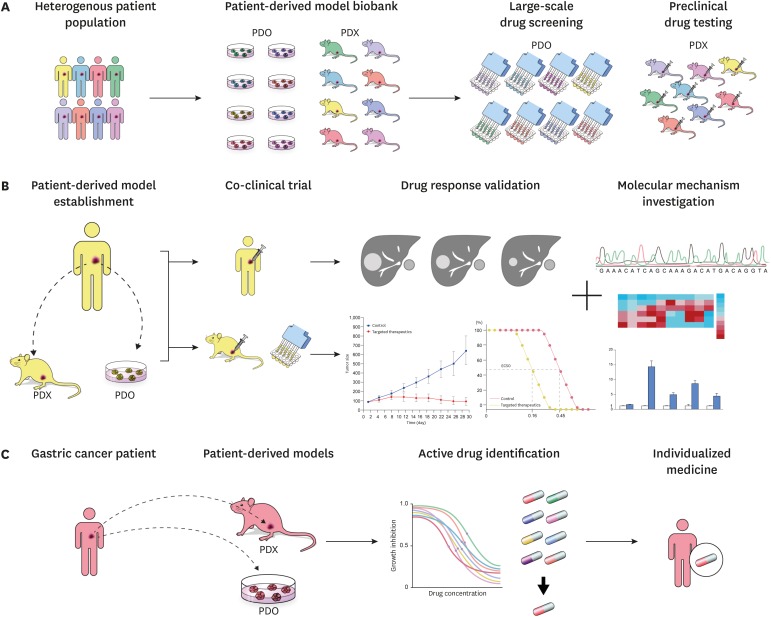 | Fig. 1Clinical perspectives of patient-derived model systems in precision medicine. (A) PDO and PDX biobanks recapitulating a heterogeneous patient population that facilitateshigh-throughput drug screening and preclinical drug testing respectively. (B) Co-clinical trials were conducted on both the patients as well as the models derived from them, enabling drug response validation and molecular mechanism investigation of response/resistance. (C) Avatar models are established from tumor samples of individual gastric cancer patients. Active drugs are identified using drug screening on avatar models, assisting physicians to optimize anti-cancer treatment.PDX = patient-derived xenograft; PDO = patient-derived organoid.
|
Although it is clear that patient-derived models will play an important role in future research and clinical practice, there still remains some challenges. Firstly, the heterogeneous nature of cancer makes it difficult to establish a standard protocol for generating and maintaining patient-derived models. For example, patient-derived GCOs have different culture requirements depending on the factors in the natural tumor microenvironment [737592]. Secondly, certain types of GC, such as diffuse type GC, have a high failure rate during PDX engraftment [42939495]. Thirdly, patient-derived models do not exhibit all the features of the parent tumor. For instance, PDOs lack blood vessels, stromal components, and an immune regulatory system, which will affect the response to treatment. Fourthly, human stroma is replaced by murine stroma in PDX models after 3 to 5 passages, and tumor fragments can only be implanted into an immunodeficient mouse [32]. Finally, it is important to continually examine the retention of key molecular characteristics of organoids and PDXs in sequential passages as genetic drift and clonal selection have been reported [328196979899]. Additional studies addressing these critical points can improve the utility of preclinical models in translational cancer research.
Go to :
FUTURE PERSPECTIVES ON IMPLEMENTING PRECISION CARE OF GC PATIENTS
Current clinical decision-making in GC depends on anatomical TNM staging. Although standardized guidelines for GC are based on evidence from large randomized controlled trials, they overlook the heterogeneous nature of cancer and recommend treatment based on the characteristics of an average population. Although efforts have been made to clarify the molecular basis for the diversity of tumor phenotypes and identify biomarkers and therapeutic targets, precision medicine for GC is still in its infancy and can be utilized only upon metastasis or recurrence.
An SPC was established for predicting the prognosis and chemotherapy response based on tissue samples from GC patients who underwent curative resection [22]. We believe that this approach is a turning point for individualized therapy in GC (Fig. 2) [100]. Subsets of patients with favorable prognosis without additional intervention after surgery can avoid unnecessary chemotherapy, whereas those who are responsive to chemotherapy will benefit from adjuvant treatment, and additional targeted drugs can be administered without delay to patients with unfavorable prognosis who are also identified as being chemotherapy resistant. Integrating clinicopathologic data, TNM staging, and SPC classification will assist clinicians in evaluating high-risk patients who have a higher probability of cancer recurrence. Furthermore, drug screening can be conducted on ‘avatar models’ of high-risk patients, proposing potential targeted drugs or rational combinations for treating cancer recurrence. By using this approach, physicians will be able to provide precision medicine to GC patients in both adjuvant and recurrent settings.
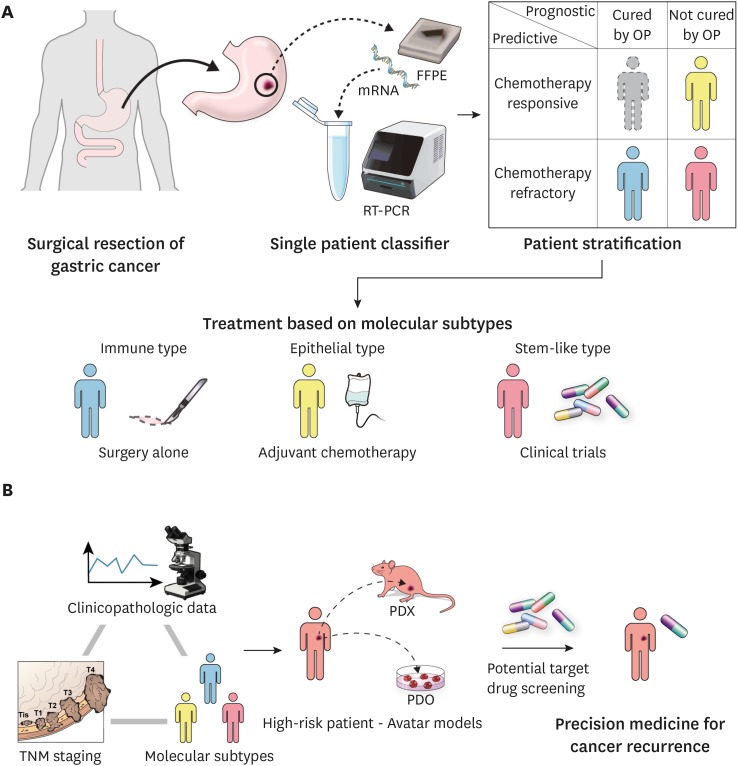 | Fig. 2Clinical implementation of precision medicine in resectable GC. (A) Resected GC samples are subjected to single patient classifier. GC patients are stratified into three subtypes: immune, epithelial, and stem-like. Optimized treatment strategy is arranged for each patient based on the clinical subtype. Immune subtypes receive no additional treatment after surgery, whereas epithelial subtypes are treated with adjuvant chemotherapy and stem-like subtypes are directly enrolled for the clinical trials. (B) High-risk patients are identified by integrating the clinicopathologic data, TNM staging, and molecular subtypes, such as high TNM stage, diffuse histology, stem-like type. Potential target drug screening is conducted on avatar models of high-risk patients to select personalized target drugs for potential cancer recurrence.mRNA = messenger RNA; RT-PCR = real-time polymerase chain reaction; GC = gastric cancer; FFPE = formalin-fixed, paraffin-embedded tumor tissue; OP = operation; TNM = tumor-node-metastasis.
|
Further investigations are needed to assess the clinical value of stratifying GC patients in an adjuvant setting. The applicability of this approach must be tested in different patient populations, such as, in GC patients in Europe where neoadjuvant chemotherapy is the preferred treatment or in American patients who have a less favorable clinical outcome than those in Asia. It is imperative to determine how to combine patient classifiers with conventional TNM staging system and treatment modalities such as targeted agents or immunotherapy. Randomized controlled trials using patient classifiers will validate this approach, whereas research using endoscopic biopsy samples will broaden its usage.
Patients who do not respond to chemotherapy and have unfavorable prognosis remain a challenge in GC treatment. These tumors are associated with diffuse histology, stem cell-like features, and EMT [2122101]. In addition, they also have a low frequency of genetic alterations and are consequently less responsive to targeted therapeutics and immunotherapy. This type of GC is characterized by slow tumor cell proliferation, high stem cell activity, mitochondria-centric metabolism, and high reactive oxygen species detoxification activity, as well as a novel synthetic metabolic vulnerability owing to nicotinic acid phosphoribosyl transferase deficiency [18102103104105106107108]. Novel drugs targeting these features warrant further examination.
Till date, only 2 targeted therapeutics, trastuzumab and ramucirumab (for anti-HER2 and VEGF2 antibodies, respectively), have been approved for the treatment of GC. Multiple clinical trials have failed to demonstrate the efficacy of other targeted agents. Most of these studies were conducted in an unselected patient population without considering the genomic characteristics of GC. Recent studies have suggested that certain agents are more effective against a specific subtype of GC [109110]. Therefore, future clinical trials should consider enrolling patients based on the presence of the target mutation.
Patient-derived models recapitulating the characteristic features of parent tumor can promote anti-cancer drug development as they serve as more precise models for preclinical efficacy tests and potential therapeutic target validation. Co-clinical trials conducted using models derived from enrolled patients will encourage researchers to develop novel strategies to potentiate the response to treatment and overcome drug resistance, while PDOs and xenografts established from resected tumor samples can function as drug validation platforms to facilitate clinical decision-making in precision medicine.
Go to :
CONCLUSIONS
GC is a genetically heterogeneous disease and each case of GC exhibits certain unique features. Elucidating the molecular characteristics of GC provides a basis for developing novel therapeutic strategies based on patient stratification. Clinical trials designed according to the molecular profiles and data from patient-derived preclinical models can lead to more personalized precision treatment for patients and better outcomes.
Go to :
 XML Download
XML Download