

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Preterm birth is the most serious problem associated with preterm labor because most babies born preterm (birth that occurs more than three weeks before the estimated due date) are at increased risks of many complications such as neurodevelopmental impairments, respiratory, and gestational complications. Thus, studies on the prevention of preterm labor and preterm birth, and the effect of drugs acting on pregnant women are important fields of study.
Many factors can contribute to preterm labor, such as vaginal bleeding, hormonal changes, stretching of the uterus (more than 1 baby, a large baby, or too much amniotic fluid) [1]. Additionally, it is well established that inflammation and/or infection represent a significant risk factor in preterm labor [2].
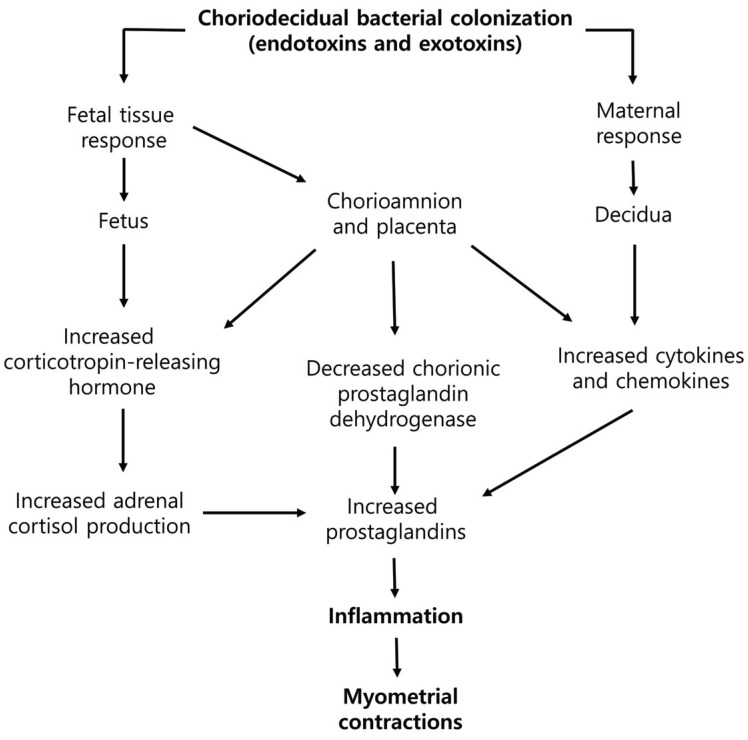
Previous studies have provided clues of how bacterial infections result in inflammation and preterm labor (Fig. 1). The first significant microbiological evidence associated with intrauterine infection before preterm birth was published in the late 1970s, when bacteria were cultured in the amniotic fluid of 7 out of 10 women who had normal membrane, but experienced preterm labor. Bacterial infections can occur within the choriodecidual space, chorion, amnion, placenta, amniotic fluid, within the umbilical cord, or the fetus [3].
Sometimes anesthesia and surgery are required to be performed during pregnancy. Each year, more than 75,000 pregnant women in the United States undergo non-obstetric surgery [4]. The operations include those directly or indirectly related to pregnancy, such as cervical cerclage, ovarian cystectomy, and appendectomy, as well as those unrelated to pregnancy, such as dental surgery. Treatment of accidental fractures or neck abscess with respiratory distress is an operation that cannot be postponed to after childbirth. Surgery can create a stressful situation and elicit an inflammatory response, and anesthetics used for pregnant women during dental surgery might induce preterm labor or birth. Therefore, it is necessary to be sure of the effects of anesthetics meant for use in pregnant patients.
Remifentanil is a synthetic opioid agonist that is used as an anesthesia adjuvant in general anesthesia. Recent studies have shown that remifentanil has anti-inflammatory effects in vitro and in vivo [56]. Remifentanil reduces inflammatory cytokine production and exerts a protective effect on lipopolysaccharide (LPS)-induced acute lung injury in rats by downregulating the nuclear factor kappa B (NF-κB) signaling pathway and acute-phase inflammatory cytokines [7]. Therefore, we aimed to determine whether remifentanil inhibits the expression of crucial mediators of LPS-induced inflammation, such as NF-κB, interlukin (IL)-1β, tumor necrosis factor (TNF)-α, and other factors using human amniotic epithelial cells (WISH cells).
METHODS
1. Cell culture
An established line of human amnion cells (WISH) was purchased from the American Type Culture Collection (ATCC® CCL25™, Manassas, VA, USA), and cultured in Eagle's Minimum Essential Medium (ATCC® 30-2003™, Manassas, VA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) in a 5% CO2 atmosphere at 37℃. Three days later, adherent cells were removed and the culture was continued by replacing the medium twice a week. We have received review exemption approval from the Pusan National University Dental Hospital Institutional Review Board.
2. Remifentanil treatment
This study used a commercially available remifentanil (GlaxoSmithKline, UK). These were diluted with culture medium, and added to cell cultures at various concentrations of remifentanil (0.001 – 1 µg/ml) with Growth medium or LPS (1 µg/ml) for 24 h.
3. MTT assay
WISH cells (1 × 105 / well) were seeded on 24-well plates and cultured for 24 h at 37℃ in a 5% CO2 incubator. After that time, cells were exposed to LPS and remifentanil (0.01 – 1 µg/ml) for 24 h. After treatment with remifentanil, the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Affymetrix, Inc. USB, OH, USA] assay was performed by adding 100 µl MTT solution (5 mg/ml in PBS at pH 7.4) into each well, and incubated at 37℃. After 1 h, the medium was removed and 100 µl dimethyl sulfoxide (DMSO; Biosesang) was added into each well. The plate was gently rotated on an orbital shaker for 15 min to completely dissolve the precipitate. The absorbance was detected at 540 nm with a microplate reader (Bio-Rad Model 680). All experiments were repeated three times.
4. Western blot
All cells were extracted with chilled RIPA buffer [50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.5% NP40, 5 mM DTT, 0.2 mM Na orthovanadate, 100 mM NaF, 1 mM PMSF] containing protease inhibitor/phosphatase inhibitor cocktail 1X (Cell Signaling Technology, Danvers, MA, USA). Samples (25 µg protein/well) were separated by Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a nitrocellulose (N.C) membrane (Whatman, USA).
The membranes were blocked in TBS-0.1% tween-20 (TBST) containing 3% skim milk for 1 h. The membranes were then incubated with α-tubulin (1:1000; Santa Cruz, CA, USA), nuclear factor kappa B (NF-κB) p65 (1:1000; Santa Cruz), Phospho-NF-κB p65 27.Ser 536 (1:500; Santa Cruz), prostaglandin E (PGE) synthase 2 (A-2) antibody (1:1000; Cell signaling Technology, Danvers, MA, USA), and cyclooxygenase (Cox) 2(D5H5) Rabbit mAb (1:1000; Santa Cruz, CA, USA), and kept overnight at 4℃ in TBST with 3% skim milk. After washing three times with TBST, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit (1:1000; Enzo Life Sciences), and anti-mouse (1:1000; Santa Cruz) for 1 h at room temperature. Then washing three times with TBST, the bands were visualized by using the ECL detection reagents (Promega, USA). α-Tubulin expression was used as the control. The target protein bands were normalized relative to the control band with an NIH Image program (Image-J launcher).
5. RNA extraction and Reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the WISH cells by using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Total mRNA (1 µg) was synthesized to cDNA using oligo (dT) PrimeScript™ 1st strand cDNA Synthesis Kits (TaKaRa Clontech, BD Biosciences, Palo Alto, CA, USA) according to the manufacturer's instructions. RT-PCR was done on a SimpliAmp Thermal Cycler (Applied Biosystems, LifeScience Technologies, CA, USA). Polymerase chain reaction (PCR) primers were as follows: interleukin (IL)-1β, Forward: 5′-CTC GCC AGT GAA ATG ATG GCT-3′, Reverse: 5′-GTC GGA GAT TCG TAG CTG GAT-3′; tumor necrosis factor (TNF)-α, Forward: 5′-CCA GGC AGT CAG ATC ATC TTC-3′, Reverse: 5′-GTT ATC TCT CAG CTC CAC GC-3′; β-actin, Forward: 5′-GAC CTG ACT GAC TAC CTC ATG-3′, Reverse: 5′-CGC TCA TTG CCA ATG GTG ATG-3′. Amplification of IL-1β/β-actin was performed using 35 cycles of 95℃ for 30 s, 55℃ for 30 s, and 72℃ for 30 s with a final extension of product at 72℃ for 7 min. Amplification of TNF-α/β-actin was performed using 35 cycles of 94℃ for 30 s, 54℃ for 30 s, and 72℃ for 30 s with a final extension of product at 72℃ for 10 min. PCR-amplified products were separated on 1.5% stained agarose gels. The Gel Doc ImageQuant LAS 500 System (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) was used to assay the PCR products. β-Actin was used to normalize all target genes. The data were analyzed using INH Image program (Image-J Launcher).
6. Enzyme-Linked immunosorbent assay (ELISA)
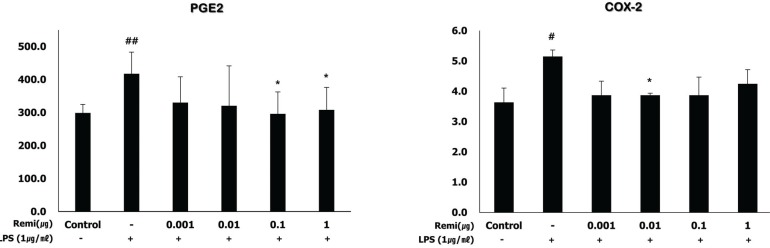
WISH cells were seeded at a density of 1 × 106 cells/ml in a 6-well plate, and incubated with remifentanil (0.001, 0.01, 0.1 and 1 µg/ml) in the presence of LPS (1 µg/ml) for 24 h following the manufacturer's instructions. Supernatants were harvested and levels of PGE2 and COX-2 were measured using a commercially available ELISA kit (product numbers ADI-900-001, ADI-900-094 respectively, all Enzo Life Sciences, Inc, USA).
The results were normalized to LPS, and presented as concentration in PGE2 and COX-2 levels of at least three independent experiments.
RESULTS
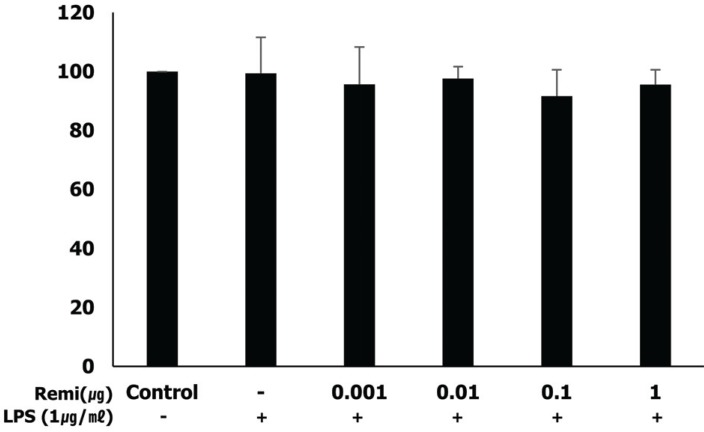
1. Effects of remifentanil and LPS on cell viability in WISH cells
MTT assays were used to evaluate the effects of remifentanil and/or LPS on WISH cells. WISH cells were incubated with remifentanil (0.001, 0.01, 0.1, and 1 µg/ml) and/or LPS (1 µg/ml) for 24 h, and then cell viability was evaluated using MTT assay. MTT assays indicated that the viability of WISH cells was not affected by the tested concentrations of remifentanil (Fig. 2).
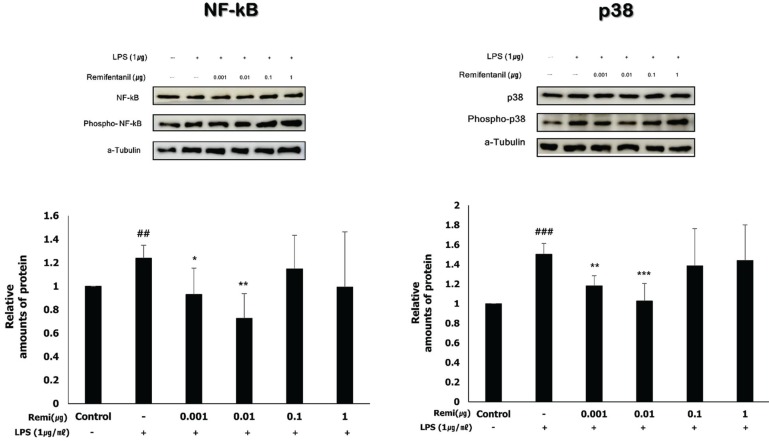
2. Remifentanil regulated the phosphorylation of NF-κB and p38 in LPS-stimulated WISH cells
Western blot analysis was performed to investigate the activation of NF-κB and p38. According to a previous study, phosphorylation of NF-κB and p38 activates the inflammatory pathway [8]. Western blot analysis suggested that LPS significantly increased NF-κB and p38 phosphorylation, and phosphor-NF-κB and phosphor-p38 levels decreased in the remifentanil co-treatment WISH cells (Fig. 3).
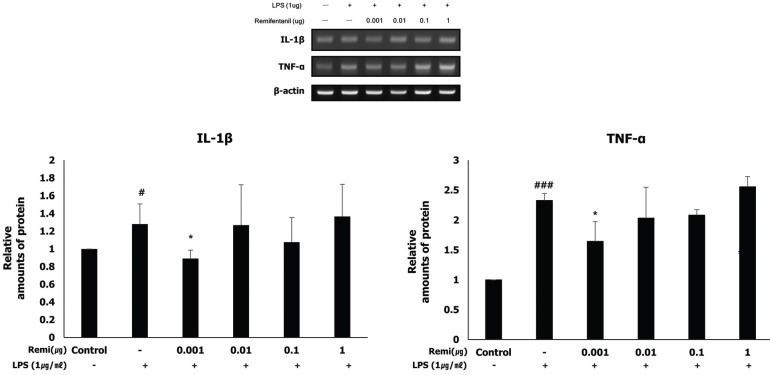
3. Remifentanil reduced the expression of pro-inflammatory cytokines (IL-1β and TNF-α) in LPS-stimulated WISH cells
RT-PCR was performed to quantify the expression of the pro-inflammatory cytokines (IL-1β and TNF-α) induced by inflammation. The analysis detected that co-treatment with remifentanil reduced mRNA expression of IL-1β and TNF-α in the cells at a very low concentration (0.001 µg) (Fig. 4).
4. Remifentanil decreased COX2 and PGE2 production in LPS-stimulated WISH cells
LPS induced cell inflammation and increased PGE2 and COX-2 levels in WISH cells. Co-treatment with remifentanil reduced PGE2 and COX-2 production after inducing inflammation with LPS in the WISH cells. It was measured using ELISA (Fig. 5).
DISCUSSION
We tested the hypothesis that premature labor may occur due to inflammatory response and that remifentanil might decrease the inflammation by downregulating various factors involved in inflammatory response, thereby protecting against early uterine contraction.
Co-treatment with remifentanil at the tested concentrations and LPS was not toxic to the WISH cells. According to previous reports, 0.5–12 ng/ml and 6–9 ng/ml blood concentrations of remifentanil were used to maintain general anesthesia for long-term use in intensive care unit sedation [910]. Thus, the remifentanil at a low concentrations (0.001, 0.01 µg/ml) in our study design are appropriate for clinical use.
LPS is the most common element within the cell wall of Gram-negative bacteria. Inflammatory cytokines are released due to its stimulation in various cell types, which provokes an inflammatory response to pathogens [11]. Toll-like receptors (TLRs) are transmembrane pattern recognition receptors (PRRs), which combine with patterns of molecular structures present on the surfaces of microorganisms. TLR-4 recognizes LPS [1213].
Stimulation of TLRs modulates the activation of NF-κ B, which leads to the production of antimicrobial peptides, cytokines, and chemokines [12]. NF-κB consists of five subunits, of which the p65 (also named RelA) and p50 subunits play a central role in NF-κB activation [14]. Phosphorylation of NF-κB occurs via two different signaling pathways, the non-canonical and canonical pathways. The canonical NF-κB pathway relies on inducible degradation of IκBα by phosphorylation, which leads to the nuclear translocation of NF-κB complexes, mainly the p50/RelA dimer [15]. From our study, cotreatment with remifentanil decreased the level of NF-κB and p38 phosphorylation at 0.001 and 0.01 µg/ml, respectively. Therefore, we can speculate that remifentanil inhibits the inflammatory response through the canonical pathway in WISH cells.
Accumulated evidence suggest that cytokines such as IL-6, IL-1β, and TNF-α play a central role in the mechanisms of infection/inflammation-induced preterm labor. IL-1 is the first cytokine involved in the onset of spontaneous preterm labor related to inflammation [161718]. IL-1 stimulates prostaglandin expression, and can stimulate myometrial contractions [1920]. TNF-α also increases the level of prostaglandin production by the amnion, decidua, and myometrium, and the concentration of TNF-α is elevated in women with preterm labor [21]. Our data demonstrate that remifentanil significantly decreased the expression of IL-1β, TNF-α, COX-2, and PGE2.
These results suggest that remifentanil may exert a cell protective effect against microbial-induced inflammatory response through the inhibition of NF-κB activation. Although this study had some limitations, we used a range of drugs that could be applied clinically, and within this drug range, we predicted that the inhibitory effect on inflammatory factors would increase with increasing drug concentrations. Remifentanil inhibited NF-κB at low concentrations and could not effectively inhibit NF-κB at concentrations higher than 0.01 µg/ml. Therefore, further research is needed to determine whether at high concentrations, remifentanil is involved in other inflammatory pathways.
 XML Download
XML Download