

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Endothelial dysfunction is the basic pathophysiology that underlies the initiation and progression of various vascular diseases such as hypertension and atherosclerosis, and ultimately leads to fatal cerebrocardiovascular events including ischemic heart diseases and stroke.12 In addition, endothelial dysfunction is known to contribute to diabetic micro- and macro-vascular complications in patients with type 2 diabetes mellitus (DM).34 Moreover, hyperglycemia is known to precipitate the development and exacerbation of diabetic vascular complications.34 In patients with type 2 DM, the endothelium is directly exposed to high glucose levels, which leads to endothelial dysfunction.5
Endothelial nitric oxide (NO) is a key regulator of endothelial cell (EC) integrity and acts as a vasodilator. Accordingly, its dysregulation is considered to contribute to the pathogenesis of vasodilation-related diseases, such as hypertension and atherosclerosis.6 The production of NO is catalyzed by endothelial NO synthase (eNOS), the activity of which is mainly controlled at the level of its phosphorylation at specific sites.78 Several of these sites, e.g., eNOS-Ser1179, eNOS-Thr497, and eNOS-Ser116, have been identified and evaluated (in bovine sequences),78 and phosphorylation of eNOS-Ser1179 has been shown to contribute most to eNOS activity and NO production.8910 Several kinases such as Akt and AMP-activated protein kinase (AMPK) have been reported to mediate eNOS phosphorylation at Ser1179,910 whereas protein phosphatase 2A (PP2A) mediates eNOS dephosphorylation at Ser1179, resulting in decreased NO production.1112
Telmisartan, an angiotensin II type 1 receptor blocker (ARB), is a first-line antihypertensive drug for the treatment of hypertension in type 2 DM patients.1314 In addition to its inhibitory effects on the renin-angiotensin-aldosterone system, telmisartan acts as a partial agonist of peroxisome proliferator-activated receptor γ (PPARγ),15 and exhibits various ancillary effects, which include cardiovascular protective effects, in a variety of cell types and tissues via PPARγ-dependent or -independent signaling pathways.15161718 Although these protective effects are observed in vitro, the protective effects of telmisartan in cerebrocardiovascular diseases are somewhat variable in the clinical trials. For example, telmisartan therapy initiated soon after ischemic stroke and continued for 2.5 years did not significantly lower recurrent stroke or major cardiovascular event rates.19 On the other hand, in another clinical trial, telmisartan modestly reduced the risk of the composite outcome of cardiovascular death, myocardial infarction, or stroke.20 eNOS-derived NO plays a pivotal role in maintenance of vascular homeostasis and its dysfunction leads to the development of various vascular diseases including hypertension, atherosclerosis, and stoke.21 Nevertheless, the direct effects of telmisartan on endothelial functions such as NO production and vessel relaxation have not been fully elucidated. Here, we investigated the molecular mechanism whereby prolonged telmisartan treatment regulates NO production and vessel relaxation in vitro and in vivo.
METHODS
Materials
Telmisartan and losartan were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Fimasartan was a gift from Boryung Pharmaceuticals (Seoul, Korea). D-glucose, D-mannitol, GW9662, acetylcholine (ACh), phenylephrine (PE), sulfanilamide, N-(1-Naphthyl)ethylenediamine, okadaic acid, and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against eNOS, p-eNOS-Ser1179, p-eNOS-Thr497, and PP2Ac were purchased from BD Transduction Laboratories (Lexington, KY, USA). Antibodies against Akt, p-Akt-Ser473, AMPK, p-AMPK-Thr172 were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against p-eNOS-Ser116 and β-actin were purchased from Abcam (Cambridge, MA, USA) and Sigma-Aldrich, respectively. Minimum essential medium (MEM) and Dulbecco's phosphate-buffered saline (DPBS) were obtained from Welgene Inc. (Gyeongsan, Korea). Fetal bovine serum (FBS), penicillin and streptomycin antibiotics, trypsin–EDTA solution, and plasticware for cell culture were purchased from Gibco-BRL (Gaithersburg, MD, USA). All other chemicals used were of the purest analytical grade available.
Cell culture and drug treatment
Bovine aortic endothelial cells (BAECs) were isolated from aortas and maintained in MEM supplemented with 5% FBS in a 5% CO2/95% air atmosphere, as described previously.1822 When BAECs had reached confluence, cells were incubated for specified times in serum-free MEM containing various concentrations of telmisartan, as described previously.18 In some experiments, BAECs were co-treated with drugs or chemicals for the indicated times.
Western blot analysis
Total proteins were extracted from BAECs or mouse aortic tissues, and subjected to western blot analyses, as described previously.18 The primary antibody dilutions used for western blot analyses were as follows; eNOS (1:2,000), p-eNOS-Ser1179 (1:2,000), p-eNOS-Thr497 (1:2,000), p-eNOS-Ser116 (1:1,000), Akt (1:1,000), p-Akt-Ser473 (1:1,000), AMPK (1:1,000), p-AMPK-Thr172 (1:1,000), PP2Ac (1:2,000), and β-actin (1:100,000).
Measurement of NO production
NO production by BAECs was measured as nitrite (a stable metabolite of NO) concentrations in cell culture supernatants and serum nitrite levels in mice were measured as total nitrite concentration, as described previously23 with minor modifications. Briefly, BAECs were treated with telmisartan in the absence or presence of various chemicals in serum-free MEM for the indicated times, and conditioned media were collected into microcentrifuge tubes and centrifuged. In a separate experiment, serum was obtained from the retro-orbital blood of mice medicated with telmisartan or vehicle. Aliquots (200 μL) of supernatant or 50 μL of serum were transferred into 96-well plates, and 50 μL of 1% sulfanilamide containing 5% phosphoric acid and 50 μL of 0.1% N-(1-naphthyl)ethylenediamine was added. After color development at room temperature for 10 minutes, absorbances were measured using a microplate reader at 520 nm. Samples were assayed in duplicate. Nitrite levels were read off a calibration curve prepared using sodium nitrite standards.
Animal studies
Male C57BL/6 mice were purchased from Orient Bio Inc. (Seoul, Korea) and housed in a temperature-controlled facility under a 12-hour light–dark cycle. Animals had unrestricted access to normal chow and water until 6 weeks of age, and were then started on a high-fat diet (HFD, D12492, 60% fat kcal; Research Diets Inc., New Brunswick, NJ, USA) for 13 weeks. Animals were then randomized to a vehicle-treated group (n = 6) or a telmisartan-treated (n = 6; 5 mg/kg body weight/day). For the next 5 weeks, animals were fed the HFD and administered telmisartan or vehicle by oral gavage. Mice were euthanized by exsanguination by retro-orbital puncture and aortas were excised and dissected. Sera were obtained by centrifuging clotted whole blood at 2,000 g for 5 minutes and aortic proteins were extracted by chopping aortas with an iris scissors in lysis buffer. Nitrite levels in sera were measured and proteins were subjected to western blotting. For ACh-induced aortic vessel relaxation assays, six-week old male Sprague-Dawley (SD) rats were purchased from KOATEC (Anseong, Korea) and acclimatized for 1 week under controlled conditions (22°C ± 1°C and RH 50% ± 10%) under a 12-hour light/dark cycle. All rats were provided with water and fed with a standard chow (Purina Mills; St. Louis, MO, USA) ad libitum throughout the experiments. On the day of assay, rats were CO2 euthanized followed by subsequent cervical dislocation, and thoracic aortas were excised and dissected.
Measurement of endothelium-dependent vessel relaxation
Endothelium-dependent vessel relaxation was measured using rat thoracic aortic rings as described previously24 with minor modifications. Briefly, male SD rats were euthanized with CO2 gas and subjected to cervical dislocation. Thoracic aortas were then rapidly and carefully removed and placed in the Krebs-Henseleit (KH) solution containing 118.1 mM NaCl, 4.7 mM KCl, 2.6 mM CaCl2, 0.6 mM MgSO4, 24.9 mM NaHCO3, 1.2 mM KH2PO4, and 5.6 mM glucose. Aorta with intact endothelium were then carefully cleaned by removing fat and connective tissues, and cut into 5-mm segments. These segments were then incubated in serum-free MEM in the absence or presence of 20 μM telmisartan in a 5% CO2 atmosphere for 24 hours at 37°C, mounted on L-shaped holders in 7 mL organ baths containing warm (37°C) oxygenated (95% O2 and 5% CO2) KH solution. Muscle forces were recorded isometrically using a force transducer (MP35; BIOPAC system Inc., Goleta, CA, USA) connected to a computer running BLS analysis software (BIOPAC system Inc.). Segments were stretched to a resting tension of 10 mN, equilibrated for 30 minutes in an organ bath containing KH solution, sequentially exposed to 65 mM KCl and KH solution at least twice, precontracted with 0.1 µM phenylephrine, and then treated with increasing concentrations of ACh to determine endothelium-dependent reactivity.
Statistical analysis
All results are represented as means ± standard deviations (SD); n values indicate the number of experiments. The significances of intergroup differences were determined using the Student's t-test for paired data. Statistical significance was accepted for P values < 0.05.
Ethics statement
All experimental procedures for a hyperglycemic mouse model were performed in accordance with the protocols issued by the Institutional Animal Care and Use Committee at the Soonchunhyang University (approval No. SCH16-0002). All experimental procedures for ACh-induced rat aortic vessel relaxation assays were conducted in accordance with the guidelines for animal care and use issued by Yeungnam University (approval No. YUMC-AEC2019-003).
RESULTS
Telmisartan decreases NO production and p-eNOS-Ser1179 levels in a time- and dose-dependent manner in BAECs exposed to hyperglycemia
Endothelial dysfunction plays an important role in the pathogenesis of various vascular diseases including hypertension and atherosclerosis, and in particular, contributes to the development of vascular complications in type 2 DM.1234 In this regard, hyperglycemia initiates the development and promotes the progression of vascular complications.34 Because eNOS-derived NO plays a pivotal role in maintenance of vascular homeostasis and its dysfunction leads to the development of various vascular diseases,625 we examined whether telmisartan affects NO production in BAECs. When normoglycemic or hyperglycemic BAECs were treated with telmisartan (0, 5, 10, or 20 μM) for 24 hours, nitrite levels (a surrogate of NO production) decreased in a dose-dependent manner (Fig. 1A). Because NO production is largely regulated by eNOS phosphorylation at specific sites78 and phosphorylation of eNOS-Ser1179 plays the most important role in the up-regulation in eNOS activity and NO production,8910 we investigated whether telmisartan affects p-eNOS-Ser1179, p-eNOS-Thr497, and p-eNOS-Ser116 levels. As was expected, treatment with telmisartan (0, 5, 10, or 20 μM) for 24 hours dose-dependently decreased p-eNOS-Ser1179 levels but did not affect total eNOS levels in normo- or hyperglycemic BAECs (Fig. 1B). In addition, telmisartan dose-dependently increased the level of p-eNOS-Thr497 but did not significantly alter that of p-eNOS-Ser116 (Supplementary Fig. 1A and B). Because telmisartan showed similar effects on NO production and the phosphorylation of eNOS in normo- and hyperglycemic BAECs, subsequent experiments were conducted under hyperglycemic conditions. Telmisartan (20 μM) decreased NO production in a time-dependent manner in BAECs (Fig. 1C), and decreased p-eNOS-Ser1179 levels when administered for 24 hours (Fig. 1D). Furthermore, treatment with 20 μM telmisartan time-dependently enhanced p-eNOS-Thr497 levels but did not affect p-eNOS-Ser116 levels (Supplementary Fig. 1C and D). These results suggest the suppression of NO production by telmisartan was caused by a decrease in p-eNOS-Ser1179 and an increase in p-eNOS-Thr497.
 | Fig. 1Tel reduces NO production and the phosphorylation of eNOS at Ser1179 in a time- and dose-dependent manner in hyperglycemic BAECs. (A) NO production was measured as nitrite (a stable metabolite of NO) in cell culture supernatants after BAECs were treated with various concentrations of Tel (0, 5, 10, or 20 μM) for 24 hours in the presence of 25 mM D-glucose or D-mannitol as described in the METHODS. (B) BAECs were treated as described above, and then p-eNOS-Ser1179 levels were assessed by western blotting. Nitrocellulose membranes were re-probed with anti-eNOS antibody to confirm equal sample loadings. (C) BAECs were treated with vehicle (DMSO) or 20 μM Tel for various times (1, 2, 4, 8, or 24 hours) in the presence of 25 mM D-glucose, and then NO production was measured as described in Fig. 1A. (D) BAECs were treated as described above, and p-eNOS-Ser1179 levels were assessed by western blotting. Nitrocellulose membranes were re-probed with anti-eNOS antibody to confirm equal sample loadings. Densitometry was used to quantitate p-eNOS-Ser1179 levels relative to total eNOS levels. All experiments were performed at least four times independently and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations below the controls (± standard deviation).Tel = telmisartan, NO = nitric oxide, eNOS = endothelial nitric oxide synthase, BAEC = bovine aortic endothelial cell, DMSO = dimethyl sulfoxide.
Differences were considered statistically significant at *P < 0.05, **P < 0.01, #P < 0.05, or ##P < 0.01.
|
Telmisartan increases PP2Ac expression, which mediates eNOS-Ser1179 dephosphorylation and consequently reduced NO production
It is well-established that several kinases such as Akt and AMPK mediate the phosphorylation of eNOS at Ser1179.910 Thus, we investigated whether these kinases are involved in the telmisartan-induced down-regulation of p-eNOS-Ser1179. Treatment with telmisartan (0, 5, 10, or 20 μM) for 24 hours dose-dependently increased p-Akt-Ser473 and p-AMPK-Thr172 levels (Supplementary Fig. 2A and B), indicating no involvement of the kinases in telmisartan-inhibited p-eNOS-Ser1179. It has been shown when eNOS-Ser1179 is dephosphorylated by PP2A, NO levels decrease,1112 and thus, we examined whether telmisartan affects expression of the catalytic subunit of PP2A, which is required for its enzymatic activity. Treatment with telmisartan (0, 5, 10, or 20 μM) for 24 hours dose-dependently increased PP2Ac expression (Fig. 2A), and treatment with 20 μM telmisartan for 24 hours increased PP2Ac expression (Fig. 2B). To confirm the role of PP2A in the telmisartan-induced down-regulations of NO production and p-eNOS-Ser1179 levels, we performed inhibitor studies using okadaic acid (a specific PP2A inhibitor). Co-treatment with 5 nM okadaic acid for 24 hours completely restored telmisartan-inhibited p-eNOS-Ser1179 levels and NO production (Fig. 2C and D). These results indicate telmisartan suppresses p-eNOS-Ser1179 levels and NO production by inducing PP2Ac expression in BAECs.
 | Fig. 2Tel increases PP2Ac expression, which mediates eNOS-Ser1179 dephosphorylation and consequently reduced NO production. (A) BAECs were treated with various concentrations of Tel (0, 5, 10, or 20 μM) for 24 hours in the presence of 25 mM D-glucose, and then PP2Ac expression levels were assessed by western blotting. Nitrocellulose membranes were re-probed with anti-β-actin antibody to confirm equal sample loading. (B) BAECs were treated with vehicle (DMSO) or 20 μM Tel for various times (1, 2, 4, 8, or 24 hours) in the presence of 25 mM D-glucose, and then PP2Ac expression levels were detected by western blotting as described above. (C) BAECs were treated with 20 μM Tel for 24 hours in the presence of 25 mM D-glucose in the absence or presence of 5 nM okadaic acid, and then p-eNOS-Ser1179 levels were assessed by western blotting as described in Fig. 1B. (D) BAECs were treated as described above, and then NO production was measured as described in Fig. 1A. Densitometry was used to quantitate PP2Ac levels relative to β-actin or p-eNOS-Ser1179 levels relative to total eNOS. Experiments were performed at least four times independently and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations above the controls (± standard deviation).DMSO = dimethyl sulfoxide, Tel = telmisartan, Oka = okadaic acid, PP2Ac = protein phosphatase 2A catalytic subunit, eNOS = endothelial nitric oxide synthase, NO = nitric oxide, BAEC = bovine aortic endothelial cell.
Differences were considered statistically significant at *P < 0.05 or **P < 0.01.
|
Telmisartan is the only ARB to reduce p-eNOS-Ser1179 and NO levels, and to induce PP2Ac expression
Although most ARBs structurally share some moieties, such as tetrazole, imidazole, and biphenyl groups, they have quite different side chains.15 Therefore, in addition to shared blood pressure-lowering effects, ARBs have specific ancillary effects, which led us to investigate whether ARBs other than telmisartan, that is, losartan and fimasartan, inhibit the phosphorylation of eNOS at Ser1179. Only telmisartan decreased p-eNOS-Ser1179 levels (Fig. 3A) and increased PP2Ac expressions (Fig. 3B). In addition, only telmisartan suppressed NO production in BAECs (Fig. 3C). These observations suggest the down-regulations of p-eNOS-Ser1179 and NO levels and the up-regulation of PP2Ac expression may be unique effects of telmisartan.
 | Fig. 3Of the angiotensin II type 1 receptor blockers tested, only telmisartan decreases the phosphorylation of eNOS-Ser1179 and NO production and induces PP2Ac expression. (A, B) BAECs were treated with telmisartan, losartan, or fimasartan (all at 20 μM) for 24 hours in the presence of 25 mM D-glucose, and then levels of p-eNOS-Ser1179 and PP2Ac expression were assessed by western blotting, as described in Fig. 2. (C) BAECs were treated as described above, and then NO production was measured as described in Fig. 1A. All experiments were independently performed at least four times, and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations above/below the controls (± standard deviation).DMSO = dimethyl sulfoxide, Tel = telmisartan, Losa = losartan, Fima = fimasartan, eNOS = endothelial nitric oxide synthase, PP2Ac = protein phosphatase 2A catalytic subunit, NO = nitric oxide, BAEC = bovine aortic endothelial cell.
Differences were considered statistically significant at *P < 0.05 or **P < 0.01.
|
Telmisartan down-regulates p-eNOS-Ser1179 and NO levels and up-regulates PP2Ac expression via a PPARγ-independent pathway
Unlike other ARBs, telmisartan acts as a partial PPARγ agonist.1526 Hence, to investigate possible PPARγ involvement in the telmisartan-induced down-regulations of p-eNOS-Ser1179 and NO levels and up-regulation of PP2Ac expression, we utilized GW9662, a specific and irreversible PPARγ inhibitor.27 Co-treatment with 5 μM GW9662 had no effect on telmisartan-induced p-eNOS-Ser1179 or NO down-regulations or on the telmisartan-induced PP2Ac up-regulation (Fig. 4). These results suggest telmisartan exerts its effects in a PPARγ-independent manner.
 | Fig. 4Tel decreases p-eNOS-Ser1179 levels and NO production and increases PP2Ac expression via a PPARγ-independent pathway. (A, B) BAECs were co-treated with 20 µM Tel and 5 µM GW9662 (a specific and irreversible PPARγ inhibitor) for 24 hours in the presence of 25 mM D-glucose, and then levels of p-eNOS-Ser1179 and PP2Ac expression were assessed by western blotting, as described in Fig. 2. (C) BAECs were treated as described above, and then NO production was measured as described in Fig. 1A. All experiments were independently performed at least four times, and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations above/below the controls (± standard deviation).DMSO = dimethyl sulfoxide, Tel = telmisartan, eNOS = endothelial nitric oxide synthase, n.s. = not significant, PP2Ac = protein phosphatase 2A catalytic subunit, NO = nitric oxide, PPARγ = peroxisome proliferator-activated receptor γ, BAEC = bovine aortic endothelial cell.
|
Telmisartan reduces p-eNOS-Ser1179 and serum nitrite levels and increases PP2Ac expression in aorta tissues and sera of HFD-fed mice
Next, to determine whether our in vitro findings were recapitulated in vivo, we performed animal studies using a hyperglycemic mouse model, which was produced by feeding mice with an HFD (60% fat kcal) for 13 weeks (Fig. 5A). In accordance with our in vitro results, the aortas of telmisartan-treated mice (5 mg/kg body weight/day) had significantly lower p-eNOS-Ser1179 levels and significantly higher PP2Ac expressions than vehicle-treated controls (Fig. 5B and C). Furthermore, telmisartan reduced serum nitrite levels by 25% as compared with vehicle controls (Fig. 5D).
 | Fig. 5Tel reduces p-eNOS-Ser1179 and serum NO levels, and enhances PP2Ac expression in the aortas and sera of HFD-fed mice. (A) Schematic diagram of animal experiments. Hyperglycemic mice were prepared by feeding male C57BL/6 mice with an HFD (60% fat kcal) for 13 weeks and then randomized to a vehicle-treated group (n = 6) or a Tel-treated group (n = 6; 5 mg/kg body weight/day), as described in the METHODS. (B, C) After being treated as described above, mice were sacrificed and aortas were dissected. Total aortic proteins were subjected to western blotting to assess p-eNOS-Ser1179 levels and PP2Ac expression (Fig. 2). (D) After mice had been treated as described in Fig. 5A, sera were obtained and nitrite levels were measured as described in the METHODS. The blots shown are representative of at least six aortas from each group. Bar graphs depict mean fold alterations above/below controls (± standard deviation).HFD = high-fat diet, Tel = telmisartan, eNOS = endothelial nitric oxide synthase, PP2Ac = protein phosphatase 2A catalytic subunit, NO = nitric oxide.
Differences were considered statistically significant at *P < 0.05.
|
Telmisartan attenuates ACh-induced aorta relaxation
To determine whether the telmisartan-regulated PP2Ac/p-eNOS-Ser1179/NO signaling axis inhibits vessel relaxation, we performed an ACh-induced vessel relaxation assay on isolated rat aortas ex vivo. Treatment with 20 µM telmisartan for 24 hours significantly attenuated ACh-induced aortic vessel relaxation versus vehicle controls (EC50 values were 17.1 μM and 0.99 μM, respectively) (Fig. 6A and B), which shows the telmisartan-regulated PP2Ac/p-eNOS-Ser1179/NO signaling axis inhibited vessel relaxation.
 | Fig. 6Tel attenuates ACh-induced aortic vessel relaxation. (A, B) Rat thoracic aortas were prepared and subjected to a vessel relaxation assay as described in the METHODS. Endothelium-intact aortic rings were treated with 20 µM Tel or vehicle (DMSO) for 24 hours in the presence of 25 mM D-glucose, precontracted with 0.1 µM PE, and then treated with increasing concentrations of acetylcholine (ACh, 0.001–10 µM). Contractile levels immediately before ACh treatment were considered to be contractions of 100%. Tension curves indicates ACh-induced aortic relaxation in response to 20 µM Tel or vehicle (DMSO). The line graph was plotted using mean ± standard deviation at each point (n = 6). All experiments were independently performed at least six times (n = 6).PE = phenylephrine, ACh = acetylcholine, DMSO = dimethyl sulfoxide, Tel = telmisartan.
Differences were considered statistically significant at *P <0.05 and **P < 0.01.
|
DISCUSSION
Clinical studies differ regarding the cerebrocardiovascular protective effects of telmisartan in hypertensive patients with or without type 2 DM. The PRoFESS study group showed that telmisartan therapy initiated soon after ischemic stroke and continued for 2.5 years did not significantly lower the rates of recurrent stroke or major cardiovascular events.19 Conversely, TRANSCEND investigators reported that telmisartan modestly reduced the risk of the composite outcome of cardiovascular death, myocardial infarction, or stroke.20 In addition, Wago et al.28 reported long-term telmisartan treatment improved endothelial function (as assessed using clinical measurements i.e., increased circulating adiponectin levels and decreased blood pressure) in hypertensive type 2 DM patients. In this regard, we also reported that telmisartan attenuated vascular inflammation by inhibiting IκB kinase β expression in hyperglycemic BAECs.1718 Additionally, we have observed telmisartan inhibits myosin light-chain kinase expression in rat vascular smooth muscle cells and aortas, and consequently attenuates PE-induced vessel contraction in rat aortas (unpublished data). eNOS-derived NO plays an important role in maintenance of vascular homeostasis by influencing platelet aggregation, leukocyte adhesion, and proliferation and migration of smooth muscle cells as well as vascular tone,25 and thus, its dysregulation contributes to various vessel-related diseases, such as hypertension and atherosclerosis.6 Nevertheless, the direct effects of telmisartan on endothelial functions such as NO production and vessel relaxation have not been fully elucidated. In the present study, we found telmisartan inhibited NO production and vessel relaxation via PP2A-mediated p-eNOS-Ser1179 dephosphorylation in vitro and in vivo in a PPARγ-independent manner (Fig. 7). Our results suggest that telmisartan exhibits inhibitory effects on NO production and vessel relaxation in ECs and aortas, apart from its known blood pressure-lowering effect caused by blockade of the renin-angiotensin-aldosterone system. Based on these clinical reports and our previous and current results, it appears the effects of telmisartan on cerebrocardiovascular diseases are dependent on the summed effects of its positive and negative actions in individual patients. In this respect, our results may explain why clinical outcomes concerning the cerebrocardiovascular protective effects of telmisartan are inconsistent.
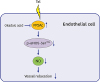 | Fig. 7A schematic illustration of Tel-inhibited NO production and aortic vessel relaxation. Tel, but not losartan or fimasartan, enhances PP2Ac expression in a PPARγ-independent manner in BAECs. PP2Ac up-regulation by Tel reduces p-eNOS-Ser1179 levels and consequently NO production in BAECs and mice. The PP2Ac/p-eNOS-Ser1179/NO signaling pathway regulated by Tel attenuates ACh-induced rat aorta relaxation.Tel = telmisartan, PP2Ac = protein phosphatase 2A catalytic subunit, eNOS = endothelial nitric oxide synthase, NO = nitric oxide, PPARγ = peroxisome proliferator-activated receptor γ, BAEC = bovine aortic endothelial cell, ACh = acetylcholine.
|
One of the most important findings of the present study was that telmisartan decreased p-eNOS-Ser1179 levels by elevating PP2Ac expression (Fig. 2), although it also induced the phosphorylations of Akt-Ser473 and AMPK-Thr172 (Supplementary Fig. 2), which are well-established kinases that mediate phosphorylation of eNOS-Ser1179.910 In particular, the enhancement of PP2Ac expression by telmisartan was entirely consistent with the phosphorylation of eNOS at Ser1179 (Figs. 1 and 2). PP2A has been shown to dephosphorylate eNOS at Ser1179 in response to ceramide,29 endostatin,30 and all-trans retinoic acid.31 Furthermore, treatment with ceramide increased the association between PP2A and eNOS, and promoted the dissociation between p-Akt-Ser473 and eNOS in BAECs, which ultimately resulted in the inhibition of p-eNOS-Ser1179.29 Vascular endothelial growth factor (VEGF)-induced phosphorylation of eNOS at Ser1177 (the human equivalent to Ser1179 in the bovine sequence) was also attenuated by treating with endostatin but did not affect VEGF-enhanced phosphorylation of Akt at Ser473 in human umbilical vein endothelial cells (HUVECs).30 Additionally, acute treatment with telmisartan was also found to simultaneously induce the phosphorylations of AMPK at Thr172 and of eNOS at Ser1177 in HUVECs, but the overexpression of a dominant-negative AMPK gene had no effect on the phosphorylation of eNOS at Ser1177.32 Based on these reports, it would appear telmisartan also inhibited the phosphorylation of eNOS at Ser1179 by inducing PP2Ac expression in the present study, regardless of the phosphorylation statuses of Ak or AMPK. However, further studies are needed to clarify the physiological relevances the up-regulations of these kinases by telmisartan.
Although most ARBs are known to share the same structural moieties, each drug possesses unique side chains.15 Unlike other ARBs, telmisartan contains a carboxyl group (rather than the common tetrazole group) linked to a biphenyl moiety and two tandemly linked benzimidazole groups,15 which suggests it probably has effects in addition to its anti-hypertensive effects. For instance, telmisartan has been shown to mitigate vascular inflammation1718 and to act as a partial PPARγ agonist.15 The biphenyl moiety and the centered benzimidazole group directly linked to this moiety have recently been reported to be essential for PPARγ activation.33 We also observed that among the ARBs tested, only telmisartan decreased p-eNOS-Ser1179 and NO levels and induced PP2Ac expression (Fig. 3). Furthermore, these effects of telmisartan were found to be mediated independently of the PPARγ pathway (Fig. 4). Based on previous reports and our results, it appears that the peculiar effects of telmisartan are stemmed from its structural properties, and particularly from the second benzimidazole group attached to the centered one. However, further investigations, including evaluation of ARBs not tested in the present study, and analyses using medicinal chemistry, are required to verify this issue.
Unlike that observed in the present study, Myojo et al.32 recently reported that acute treatment with telmisartan activates eNOS via the p38-mediated phosphorylation of eNOS at Ser1177 in HUVECs. It was found that treatment with 10 μM telmisartan for 2 hours maximally increased p-eNOS-Ser1177 levels. Because telmisartan is administered as an anti-hypertensive orally once daily and its elimination half-life is approximately 24 hours,34 treatment with telmisartan for a relatively long period of time (24 hours) more closely mimics the clinical situation. Furthermore, sequences around eNOS-Ser1179 constitute a substrate motif for AGC kinases such as protein kinase A, protein kinase G, and protein kinase C, whereas p38 is a CMGC kinase family member, namely cyclin-dependent kinase (CDK), mitogen-activated protein kinase, glycogen synthase kinase, and CDK-like kinase.35 Therefore, it is unlikely that p38 is involved in the direct eNOS phosphorylation at Ser1177. However, we cannot fully explain the inconsistency between previous report and our findings, though it may be due to differences between the experimental conditions (e.g., telmisartan concentrations and treatment times) and the cell types used. Nonetheless, the present study showed that telmisartan reduced the phosphorylation of eNOS at Ser1179 by up-regulating PP2Ac expression in vitro and in vivo, and our ACh-induced ex vivo aortic relaxation assay observations demonstrated physiological relevance (Figs. 2, 5 and 6).
High interpatient variabilities in telmisartan plasma concentrations have been shown in patients with mild to moderate hypertension; mean +/− SD values for Cmax were 159 +/− 104 ng/mL for 40 mg telmisartan, 693 +/− 606 ng/mL for 80 mg telmisartan, and 1,635 +/− 1,406 ng/mL for 120 mg telmisartan (equivalent to 0.31 +/− 0.20 μM, 1.35 +/− 1.12 μM, and 3.12 +/− 2.73 μM, respectively).36 Additionally, Zhang et al.37 reported mean +/− SD values for Cmax in healthy Chinese subjects to be 163.2 +/− 128.4 ng/ml and 905.7 +/− 583.4 ng/mL for 40 mg and 80 mg telmisartan (equivalent to 0.32 +/− 0.25 μM and 1.76 +/− 1.13 μM, respectively). Furthermore, Cmax was 3,200 ng/mL for 160 mg telmisartan, which is equivalent to 6.11 μM.34 If the telmisartan concentrations used in our in vitro experiments (5–20 μM) are compared directly with these clinical data, our concentrations would appear to be higher. Nevertheless, in the present study, telmisartan (10 μM) enhanced PP2Ac expression and suppressed the phosphorylation of eNOS at Ser1179 and NO production, although greatest effects were observed at a telmisartan concentration of 20 μM (Figs. 1 and 2). Moreover, higher telmisartan concentrations (100 μM) have been used in other in vitro studies.3839 Thus, we consider the concentration of telmisartan used in our in vitro study is reasonably compatible with clinically observed peak Cmax values.
Summarizing, the present study demonstrates telmisartan inhibits NO production and vessel relaxation at least in part by the PP2A-mediated dephosphorylation of eNOS-Ser1179 in vitro and in vivo in a PPARγ-independent manner. These results may provide details of the underlying mechanism responsible for reported inconsistent clinical outcomes regarding the cerebrocardiovascular protective effects of telmisartan.
 XML Download
XML Download