

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
A recent consensus document [1] recognized the need to advance in precision medicine in allergic diseases as well as to “improve the process of drug development, biomarkers and companion diagnostics for allergic diseases and asthma.”
Looking for adequate biomarkers to stratify allergic patients has been a subject of active research in the last decades [2], but finding these biomarkers is not easy. Potential candidate biomarkers should be supported by a body of evidence, meaning that they should have a biological explanation. At the same time, they should be quantifiable in a biological matrix in an efficient, cost-effective and reproducible analytical technique. Ideally, the biological matrix should be obtained in an easy, quick and efficient way, such as serum or plasma.
Historically, allergy biomarker research has been focused on antigen-specific biomarkers, such as specific serological or cellular biomarkers. In the last years this approach has been progressively enriched with new biomarkers connected to inflammatory status. Some of these new biomarkers have already been incorporated in clinical practice (i.e., periostin or fraction of exhaled nitric oxide).
In parallel to this, in the last years there has been an active development of increasingly effective new pharmaceuticals for the control of symptoms in mild allergic patients, as well as a continuous development of new biological drugs tailored to control severe allergic phenotypes. The elevated cost of these therapies has fuelled the search of accurate definitions of disease endo-phenotypes. Etiological allergy management and associated allergen-specific immunotherapy (AIT) is positioned between the 2 above-mentioned pharmacological intervention strategies being the only pharmacological approach with demonstrated evidence of disease-modifying effect [34]. The problem to prove the value of etiological management is connected to the absence of adequate biomarkers to predict and monitor effect on a patient-by-patient basis and to the lack of common biomarkers that would allow head-to-head comparison of different pharmacological approaches. These types of biomarkers would facilitate the monitoring of residual effect after therapy cessation, where AIT intervention should have the highest therapeutic value. In this context, omics methods in general—and specifically metabolomics—provide new tools to develop a rational approach to the development of new biomarker strategies.
OMICS SCIENCES IN ALLERGY
Omics sciences have significantly contributed to the definition of new biomarkers. These sciences are based on the use of high amounts of data and bioinformatic high-throughput techniques. Moreover, in the last decades the field of omics sciences has experimented huge technological advances by improving detection limits and developing software tools for the analysis and visualization of data.
Omics sciences include genomics, transcriptomics, proteomics and metabolomics among others (Fig. 1). Genomics is focused on the structure, function, evolutionary mapping and editing of genomes, and defines the potential genetic features of a person which are associated with a disease. As an example, several polymorphisms have been studied in different allergic diseases. That is the case for a glutathione transferase polymorphism that has been associated with asthma risk [567]. Transcriptomics, on the other hand, is the science that studies the sum of all RNA transcripts in any of their forms, including messenger RNA, ribosomal RNA, transfer RNA, microRNA and other noncoding RNAs. In fact, microRNA analysis has been extensively used in the search of new biomarkers in asthma [8]. Moreover, an important feature of transcriptomes is that they are different among cell populations and vary with environmental conditions, providing a broad information of which cellular processes might be occurring in a specific condition or disease. Proteomics is the characterization of structure, function, interaction or modification of proteins at any stage. As occurs in transcriptomics, the proteome also fluctuates depending on the moment, cell type and surrounding environment, and reflects the mediators responsible for a specific mechanism involved in a disease onset and/or progression. This methodology has been extensively used in the identification and characterization of novel allergens [9] and in the elucidation of novel biomarkers associated to allergic diseases, as is the case of allergic dermatitis [10]. Finally, metabolomics is the science that studies the metabolome, which is constituted by the intermediate and final molecules of the metabolism. Therefore, metabolomics reflects the exact processes that are taking place associated to disease progression. Our group has applied this technique in the search of novel biomarkers associated with severe allergic and asthmatic profiles [1112].

All the above-mentioned omics sciences are relevant in the identification of novel biomarkers in allergic diseases and asthma. Omics data are generated by high-throughput biotechnological platforms delivering hundreds of thousands of raw (nonelaborated) variables that have revolutionized medical research. Most of the studies published currently are using a single omic science to characterize biological features. However, the potency of omics analyses will significantly increase if we are able to integrate several of them to generate a “complete picture” of what is happening in a specific subject. Moreover, another dimension of data integration is between omics and nonomics data, which will bring the omics results closer to the daily clinical practice. This integration provides the opportunity to get insights into biological systems of health and disease, in order to conduct translational and personalized medicine.
METABOLOMICS
Although metabolomics was the latest omic science to be consolidated, it is indeed the oldest of the omics. It was in the time of ancient Greece where “urine charts” were used as a diagnostic tool. These charts contained different aspects of urine such as color, smell and/or taste, which used to be linked with the diagnosis of various medical conditions [13]. At the present, the diagnosis of many diseases is performed based on specific compounds from the metabolism. The molecules that cover the entire metabolome are called metabolites. The classical example of a metabolite that currently acts as a biomarker in a disease is glucose for the diagnosis of diabetes.
The metabolome from a living organism is not only composed of its own metabolites, but also of the metabolites coming from the microbiome—especially from the gut—[14], the metabolites from xenobiotics—for example those coming from medicines—, and the metabolites from the diet [15]. The levels of metabolites in the organism are conditioned by different factors such as the level of exercise, age, gender, living-stress and the environment [1617].
The introduction of high-throughput analytical techniques for metabolomics has allowed the development of many different strategies to explore the metabolome in a disease. There are 2 main complementary approaches for working in metabolomics. These are called nontargeted and targeted analyses (Fig. 2) [1819].

Regarding nontargeted analysis, this approach aims to measure as many metabolites as possible in a single run. These metabolites measured together from a single sample are considered as the metabolic fingerprint of the patient. Therefore, it is estimated that in a disease status there is a set of metabolites that will change due to the progression of the pathology and will characterize the disease. Thus, once the metabolic fingerprints are obtained, the aim of metabolomics is to study the significant differences in the metabolite relative abundances between groups. Consequently, this type of approach does not need previous knowledge or a specific hypothesis other than that the groups in the study are distinct. Therefore, through this strategy new potential biomarkers of a clinical condition or phenotype can be obtained. This interesting approach is strengthened if multiple high-throughput analytical techniques are applied to the same sample, since each of them gives complementary information about the metabolome.
On the other hand, targeted analysis focuses on an accurate quantitation of a limited set of metabolites. This type of approach is closer to classical biochemistry, where metabolites are measured and quantified following previous knowledge that they may be dysregulated. The innovation in respect to classical biochemistry relies on the introduction of new instrumentation which allows the determination of this set of compounds in a single run with very high sensitivity.
Both types of approaches can be used independently; however, in new topics such as finding biomarkers in severe allergic phenotypes where the metabolic characterization has not yet been performed, the first step would be a nontarget analysis. Therefore, a broad scheme of the metabolomics workflow to consolidate a trustworthy panel of biomarkers is first the “generation of a hypothesis” as a starting point to set up what is happening in the metabolism of the severe allergic group, then the “analytical validation” of the potential biomarkers and finally the “clinical validation.” In the “generation of the hypothesis” phase a nontargeted analysis is carried out. This is usually done with a small population but with a strict and homogeneous clinical phenotype [2021]. The small number is usually used because the samples must be analyzed at the same time, and the analytical techniques used may struggle to provide reliable and reproducible data if the number of samples is too high. Together with the high cost of analysis and the massive amount of data obtained per sample, small numbers of patients are chosen, such as twenty per group. However, it is not always possible to select a limited number of subject with a perfect clinical stratification, and thus tremendous work has been put in order to succeed in the analysis of large-scale cohorts with lower classification accuracy [22]. Later on, for “analytical validation” stage, targeted analysis is generally used. In this step, the quantitation of a set of potential biomarkers is done in bigger cohorts, around hundreds of samples. In this case, where the analysis is accurate, the clinical phenotyping does not need to be strictly homogenous. Finally, in the “clinical validation” stage, thousands of samples are analyzed before establishing a set of biomarkers for a disease.
NONTARGETED ANALYSIS IN METABOLOMICS
As the nontarget analysis is the hypothesis-generating step, it is important to understand the steps that take place in order to find potential biomarkers accurately. The nontarget analysis workflow can be divided in the following stages: sample collection, metabolite extraction and sample analysis, data processing, statistical analysis and biological interpretation.
Every stage of the nontarget workflow is crucial to obtain reliable findings. In the case of sample collection, it is essential—in the case of blood—to take the sample from the patients in fasting conditions, usually 8 hours minimum [23]. Moreover, serum and plasma must not be mixed, so it is important to establish which type of blood sample will be used in the project from the beginning. Besides, apart from other differences, serum is the result of a coagulation process whereas plasma is the raw matrix from the system with only the cells removed. Apart from blood, other biological samples that can be analyzed by metabolomics include urine, faeces, exhaled breath, sputum, and so on. However, each of them (including blood) gives different and complementary information; for example, in urine, only the ending compounds of the metabolism are found, although it is widely used due to the ease of the collection process [2425].
The metabolite extraction is carried out depending on the analytical technique that will later be used. There are some standards protocols to treat the samples for metabolomics. However, common to all, the interferences—mainly DNA, RNA and proteins—are removed before the analysis. This is usually done by mixing the sample with organic solvents such as methanol or acetonitrile. In addition, it is possible to choose a specific group of metabolites—for example lipids—where the metabolite extraction should be done with highly non-polar solvents such as chloroform or ether. Moreover, specific analytical techniques require a chemical modification of the metabolites in order to be measured.
Regarding the high-throughput analytical techniques in metabolomics, these are mostly based on 2 techniques: nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS)-based techniques [26]. This fact is because essentially both give structural information of the metabolites. Therefore, once the significant metabolites are selected, they can be identified and thus shed some light about the disrupted metabolic processes. In addition, there are other types of devices which are used specifically in respiratory diseases, which is the case of the electronic nose. These devices use sensor arrays which produce an electronic signal when they get in contact with metabolites from the exhaled breath of a patient. Their use in some pathologies such as asthma or allergy has shown promising results [2728]. However, it is important to take into account that these devices do not provide structural information of the metabolites; thus, they are being used for diagnosis but not as a tool to understand the mechanisms involved in the disease.
In the case of NMR and MS-based techniques, each analytical technique has strengths and drawbacks, and provides unique and complementary information of the metabolome. Regarding NMR spectroscopy, this technique detects the signals from metabolites possessing a specific type of atoms, such as hydrogen, and therefore it is abbreviated as 1H NMR. Thus, this technique allows the determination of all metabolites which contain hydrogen and are in high concentration. This technique has gained great attention because it is nondestructive, robust and reproducible. Moreover, NMR has the advantage of obtaining an accurate structural elucidation through the application of 2-dimensional spectra analysis. However, its major drawbacks are its low sensitivity and the difficulty of treating and interpreting the complex data profiles obtained [29].
On the other hand, MS-based techniques have gained great popularity due to their high sensitivity. They are based on measuring the molecular mass of the metabolites, specifically their mass-to-charge ratios, which are symbolized as m/z. This is possible only if the metabolites are ionized or charged before entering the mass spectrometer. One of the most used ionization sources is the electrospray ionization, by which metabolites gain a charge without being fragmented. The ions pass to the mass analyzer, which detects the mass of the metabolites with high mass accuracy. A common example of mass analyzer is the time of flight (TOF)-MS, which provides a mass resolution of up to a thousandth of an atomic unit (or Da). Furthermore, MS-based techniques have the great advantage that they are usually coupled to a separation technique such as liquid or gas chromatography (LC or GC, respectively), or capillary electrophoresis. By this coupling, the metabolites in complex samples—such as blood or urine—are first separated, and then analyzed with high mass accuracy. The separation adds value to the later identification of significant metabolites as the retention time gives information of the physicochemical properties of the metabolites. As an example, it is not expected to observe a triglyceride at the beginning of a chromatogram obtained from a reverse phase chromatography, as the compounds in this zone should be very polar.
In the case of LC-MS, this is maybe the most widely used technique for metabolomics. This technique combines the suitability to analyze a wide range of metabolite classes— including lipids, metabolites with intermediate polarity and polar compounds—with a high sensitivity and with the possibility of doing compound fragmentation [30]. The fragmentation of a compound is almost compulsory to obtain a reliable identification of a metabolite. The only requirement to use LC-MS is to have the sample in liquid form before analysis. Among the drawbacks of the technique is the low stability during large-scale analyses, although great improvements have been done in the protocols in the last years [22].
On the other hand, GC-MS is a technique that allows the measurement of volatile compounds, and thus, it is suitable for the analysis of breath. However, the most common matrices analyzed by this technique [31] are blood and urine, as there are well-established protocols for them [32]. This is possible due to a chemical process called derivatization, which provides volatility and stability to metabolites contained in complex biological samples. Among the types of metabolites that can be measured with this technique are: amino acids, sugars (pentose and hexose sugars), disaccharides, metabolites from the Krebs cycle, organic acids, fatty acids and cholesterol. The main drawback of GC-MS is the extensive sample preparation needed for nonvolatile metabolites. One of its best advantages is that the identification is done based on specific GC-MS spectral libraries before the statistical analysis.
Regarding CE-MS, this is an excellent technique to analysze polar metabolites from aqueous samples such as urine. However, as in GC-MS, well-established protocols have been developed to analyze other biofluids, such as blood or tissue [33]. Among its strengths, the main one is the small amount of sample (few nanolitres) and reagents needed. On the other hand, its main drawback is the lower sensitivity compared to LC-MS due to the low volume of sample.
In the case of all MS-based techniques, it is compulsory to measure a quality control (QC) sample in order to assess the reproducibility and stability of the technique. This QC is usually done by pooling a small aliquot of each sample in the study. In this way, the QC is a representative sample of the study and it has to be injected at regular intervals (usually every 5 samples) throughout the sequence to ensure the stability of the technique. It is important to highlight that the use of more than one analytical platform in a study provides a higher coverage of metabolites and therefore a better “picture” of the metabolic status of the patient.
Once the sample analysis is finished, the data from the metabolic profile are extracted, which usually results in huge amounts of variables (or metabolites) per patient. This aspect entails that the statistical analysis of the data must rely not only in univariate but also commonly in multivariate analysis. The univariate analysis applies the traditional statistical tests—such as t test—to each variable to test their significance. Along with the test, a correction of the p value is performed to control the false discovery rate. On the other hand, in the multivariate statistical analysis, all variables are used to model the differences between groups. There are 2 types of statistical models, unsupervised models such as the “Principal Component Analysis” (PCA) and the supervised model called “partial least square – discriminant analysis” (PLS-DA). In the PCA, the software does not count with any information of the samples and using the Nipals algorithm displays a graphic with the location of each sample according to their similarity with each other. The PCA model is important for looking at sample trends and patterns, and therefore it is usually used to detect strong outliers. In the case of the PLS-DA, the model looks for the variables that can discriminate between the groups of the study. Therefore, this model is used for sample classification and for prediction of a new sample. To sum up the statistical analysis part, usually the selection of the significant metabolites between the groups of study is done based on univariate and multivariate analyses [3435].
Furthermore, once the metabolites that discriminate between 2 or more groups are selected, these have to be fully identified. As was said before, this process varies depending on the analytical technique used. Once a list of differentially regulated metabolites is obtained, it is important to explain their molecular mechanism. This is usually done based on the analysis of the metabolic pathways, which are ranked according to their enrichment and impact parameters. The enrichment of a pathway means the number of metabolites found in a specific pathway while the impact refers to if the metabolites are specific for a metabolic pathway or are shared with other pathways. Of course, the metabolic pathways are not completely known and because of that it is important to consult all possible scientific literature about these metabolites in order to get the best interpretation. Furthermore, the metabolites discovered and their possible explanation can be corroborated on new biological models such as mice or cell models. On the other hand, these potential biomarkers can be moved to the next step of the “analytical validation” and be tested in hundreds of new samples. This step would use the target analysis approach of metabolomics and would be based on receiver operating characteristic (ROC) curves where the best metabolite predictors will be selected. However, usually the research projects stop once the nontarget analysis finishes because of the amount of time that takes this step (around 2–3 years) and the complexity for collecting new samples and developing analytical methods in target.
The work in metabolomics is often challenging and implies the collaboration of multidisciplinary specialists (medical doctors, chemists, biologists, statisticians, bioinformaticians, etc.); however, the result of finding new biomarkers in severe allergic phenotypes will definitely improve their diagnosis and their medical treatment.
TARGETED ANALYSIS – ANALYTICAL VALIDATION
The “analytical validation” stage using targeted analysis starts with a list of potential biomarkers obtained from a nontargeted analysis. The limitation of the targeted analysis starts with the fact that not all the significant metabolites have a commercial standard. This fact reduces the number of compounds to be later tested in the targeted analysis.
Furthermore, for this type of approaches the analytical technique of choice is LC-MS, but in this case with a mass analyzer called triple quadrupole (QqQ). A quadrupole, as its name suggests, consists of 4 parallel metal rods arranged in a square. The purpose of the quadrupole is to separate the charged metabolites—or any kind of ions—according to their m/z values by applying a range of specific radiofrequency electric fields and voltages. This means that a specific mass can be selected if specific values of radiofrequency and voltage are set. In contrast to TOF mass analyzers, the quadrupole has low mass accuracy, usually up to the first decimal of an atomic unit (or Da). However, in order to compensate its low resolution, 3 quadrupoles are used and arranged one after each other. This type of configuration is called QqQ. Each quadrupole has a distinct function: the first quadrupole selects a mass (no matter the low mass resolution), the second quadrupole acts as a collision cell where the charged mass (and compounds with close masses) are fragmented, and the third quadrupole isolates a mass from a fragment. The selection of an initial mass which gives a specific fragment is called transition and is considered unique of the compound or metabolite. Moreover, thanks to this method of analysis, the QqQ-MS instrument possesses one of the highest sensitivity and selectivity capacities [36].
Therefore, in order to apply a targeted analysis, an analytical method development should be carried out. As any other analytical method, this must be validated at least in linearity, precision, accuracy and limit of detection and quantitation. Moreover, this type of analysis usually needs labelled standards. These are molecules with the same formula as the metabolites where a hydrogen or a carbon atom is replaced by deuterium or carbon thirteen, respectively. These types of compounds are usually used with the aim of controlling the stability of the instrument and the analysis. Moreover, there are specific metabolites that will be charged only with one of the 2 modes of ionization. However, working with QqQ-MS, the change from one ionization to another during the analysis of one sample is possible.
One of the advantages of targeted analysis is that the samples can be measured at different time points and later the results can be joined and compared between batches. In order to establish the most robust metabolites from the list of potential biomarkers, a set of different statistical analyses should be done. Among these, the ROC curve using a combination of potential biomarkers provides a straightforward option [37]. The goal is to establish the combination of biomarkers that shows the highest specificity and sensitivity for detecting patients with a severe allergic phenotype.
SEVERE PHENOTYPES IN ALLERGY TO UNDERSTAND DISEASE PROGRESSION
Severe phenotypes in allergy disease constitute a small fraction of overall disease burden. However, with the development of new biological drugs, this small fraction of patients is attracting an increasing number of pharmaceutical developments due to their high pharmaco-economic potential. Usually, these types of patients are randomly distributed within the overall population of allergic patients and are difficult to identify.
When applying metabolomics strategies to discover new biomarkers, it is essential to include patients with different stages of disease progression in a homogeneous way. However, there is no easy way of doing this. First, allergic patients are progressively treated with different drugs aiming towards disease control. This control will possibly alter systemic signatures associated to progression of the disease and will hide the underlying endo-phenotype.
Uncontrolled severe patients, meaning patients that are not well controlled with any combination of existing pharmaceutical products, represent a unique group that will be pivotal to understand disease evolution and to identify systemic molecular signatures associated to disease evolution.
Disease progression is linked to different potential factors. It is generally accepted that genetic factors and exposome [38] will determine the probability to develop a severe allergic phenotype. However, it is difficult to evaluate the genetic influence in allergy due to the inadequate design of many published studies and the complexity of multifactorial effects of affected polymorphisms [39]. Regarding exposome, it has been described that patients exposed to high allergen levels are more prone to develop a more severe allergy disease. In an epidemiological survey based on molecular allergy, it was described that patients overexposed to grass pollen are frequently sensitized to profilin. In the same study, those patients overexposed to olive pollen presented a significant increase in the sensitization prevalence to minor allergens such as Ole e 7 [4041]. Interestingly, it was observed that a significant fraction of patients resident in overexposed areas that presented these sensitization profiles develop a more severe phenotype [4243]. Likewise, in an area of high mite allergen exposure, as is the subtropical climate region of Canary Islands in Spain, there are patients that develop anaphylaxis reactions when eating mite-contaminated foods in the absence of food allergy [44].
These 3 groups of patients present several clinical features connected to severity (asthma, severe anaphylactic reactions…) and do not respond to AIT. We thus decided to investigate using omics and nonomics approaches if there are common features that could explain the underlying causes for severity evolution.
NOVEL MECHANISMS ASSOCIATED WITH SEVERE ALLERGIC PHENOTYPES
We started by studying the profilin/grass pollen allergy model. We discovered that in the progression of grass pollen/food allergy into a more severe phenotype, oral mucosa plays a pivotal role [45]. By using an oral provocation clinical model with pure profilin on severe food/grass pollen allergic patients, we demonstrated that oral mucosa is damaged in severe allergic patients. Our results showed a significant reduction of tight junction proteins such as claudin-1 and occludin and e-cadherin, protein member of the adherens junctions. Moreover, epithelial damage in severe patients was also reflected by a significant downregulation of interleukin-33 and Periostin in the oral mucosa. In this model, oral mucosa disruption is accompanied by infiltration of a significant number of inflammatory cells (CD11+ and CD3+ cells), suggesting also a local inflammatory response in the oral mucosa [45]. Interestingly, no eosinophils or neutrophils were detected. These results were the first demonstration of the role of oral mucosa in the onset of systemic reactions, but they also opened up a question. Was the observed remodeling a consequence of the food allergy phenotype, or was it produced earlier connected to respiratory allergy and later evolved to a food hypersensitivity?
To answer these questions, we studied severe allergic patients sensitized to olive pollen and mites, as previously described. The results showed that in the oral mucosa of these patients, which were exposed to high levels of olive pollen of house dust mites (HDM) allergens and presented no food allergies, oral mucosa integrity was also disrupted. However, these patients do not present inflammatory infiltrates in the oral mucosa, probably due to the lack of oral antigen exposure. Altogether, these data suggested that in severe respiratory allergic phenotypes, there is a systemic exacerbated inflammatory response underlying the disruption of the oral mucosa integrity [46].
From these results, we can infer that severe allergic patients might present a specific modulation of biological pathways associated to the underlying inflammatory response. In fact, those specific pathways and associated molecules can be identified as potential biomarkers of severe allergic phenotypes by using omics technologies.
To better understand the severe-phenotype-associated mechanisms using the same model of profilin severe food-associated respiratory allergy, transcriptomic and metabolomic analyses were performed in peripheral blood mononuclear cells and plasma, respectively [12].
Transcriptomic analysis revealed a significant downregulation of platelet-associated genes in the severe phenotype. Gene expression and gene pathway analysis revealed downregulation of platelet activation, secretion, adhesion and aggregation [12]. These data, although surprising at first, agreed with previous results found in literature, where platelets appear to have an important role in allergic diseases and asthma [47].
Moreover, the metabolomic analysis also revealed significant differences in the severe group regarding several metabolic pathways. The energetic metabolism was modified, as severe allergic patients presented high levels of lactic acid and low levels of pyruvic acid, which correspond with a metabolic phenomenon called Warburg effect, common in cancer progression. This metabolic feature could be associated with a higher activation of T cells during a systemic inflammatory response [48].
Another important metabolic modification found in this severe allergic phenotype was the increase of lysophospholipids (Fig. 3). The metabolites called LPC 16:0 and LPC 18:0, which were increased in the severe allergic group, are the products of the Phospholipase A 2 enzyme. These metabolites are precursors of ether-linked phospholipids, which are intermediates in the synthesis of the platelet activation factor, PAF [49]. In addition, they are a source for the synthesis of eicosanoids such as arachidonic acid and derivatives [50]. Finally, LPC 16:0 is a source of palmitic acid, which can be in turn used for the synthesis of sphingosine, a precursor of sphingosine-1-phosphate [51]. Moreover, serum increased levels of lysophospholipids have been previously described in asthmatic patients [50]. Interestingly, patients sensitized to profilin in areas of higher grass pollen exposure showed an enhanced T-cell proliferative response to profilin, higher than in patients sensitized to profilin in areas of relatively lower grass pollen exposure, supporting the role of T-cell proliferation in the onset of severe respiratory phenotypes [52].
Fig. 3
Schematic representation of biological pathways associated with lysophospholipids in inflammation. PAF, platelet activation factor.

In addition, we detected sphingolipid metabolism stress in severe allergic patients [12]. More specifically, a continuous increase in sphingosine 1 phosphate (S1P) was accompanied by signals of the exhaustion of metabolic precursors [53].
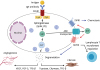
S1P has been described as an essential player in the immune response [54] (Fig. 4). It is involved in the activation and degranulation of mast cells and associated induction of angiogenesis and fibrosis triggered by endothelial and epithelial cells [55]. This metabolite also plays an important role in T-cell biology by increasing T-cell lifespan and the recruitment of T cells to the site of injury [56], as has been reported in lung diseases models [57]. Altogether, the data suggest that a global system connected to inflammation is pushed to the limit in severe patients. In this system, sphingosine metabolism, T-cell proliferation and platelet functionality are involved.
Fig. 4
Role of sphingosine 1 phosphate in mast cells. Potential implications in angiogenesis, fibrosis, and T-cell biology. FCER1, high-affinity IgE receptor; S1PR1, sphingosine 1 phosphate receptor 1; S1PR2, sphingosine 1 phosphate receptor 2; ABC, ATP-binding cassette; VEGF, vascular endothelial growth factor; FGF-2, fibroblast growth factor 2; TGF, transforming growth factor.
We have used a similar approach in an HDM-allergic asthma model aiming to identify novel biomarkers or validate previous biomarkers associated with other severe allergic models. Initial unpublished generated data using nontargeted metabolomic analysis of serum samples demonstrates that, once more, uncontrolled asthmatic patients display a specific metabolic fingerprint, completely different from controlled asthmatic patients. Most of metabolomic signatures are shared with our previous severe allergy models, further supporting that severe allergy patients share underlying mechanisms. Therefore, the use of metabolomics can be considered a useful and potent tool in the identification of novel biomarkers of severity in allergic diseases and asthma.
FUTURE DIRECTIONS
Understanding underlying biological processes in allergy evolution is pivotal for a correct interpretation of new biomarkers arising from ongoing studies.
Allergy is a complex disease and severe phenotypes often develop in association with different comorbidities.
Data arising from these metabolomic studies depict an image with multiple interconnected systems. Barrier impairment, innate immune system activation and progressive inflammation are central to understand allergy. There are new players in the game. Platelets seem to potentially play a relevant role in the maintenance of an inflammation/repair balance. In this balance, sphingolipid metabolism also seems to play an important role. Almost half of detected metabolic signatures are connected to phospholipid consumption associated to main inflammatory routes. Moreover, altered energy metabolism connected to T-cell proliferation points to the need of focusing on T regulation as the main target in allergy clinical management, a target that currently is unique for AIT intervention.
Metabolomics is providing increasing value for understanding underlying causes associated to severity in allergy; however, there is still a long way to go. We need more severity models and to compare the different information provided by each one.
Existing data support that an initial panel of around fifty metabolites is a potential candidate to stratify clinically allergic patients. Target method development for these metabolites is underway, and exploratory projects with thousands of patients to analyze the value of such a panel are being initiated.
 XML Download
XML Download