

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Citalopram is a commonly used selective serotonin reuptake inhibitor (SSRI) to treat major depressive disorders [1]. However, citalopram is a racemic mixture of R and S-enantiomer form. Since S-enantiomer is more active than R-enantiomer, escitalopram, an S-enantiomer of the citalopram, has been developed. It shows markedly higher potency than citalopram for treating depression and anxiety disorder [2]. This could be explained by allosteric binding of escitalopram with serotonin reuptake transporter which enhances binding affinity of escitalopram at the primary binding sites, resulting in more effective blocking of 5-hydroxytryptamine (5-HT) reuptake [3].
Recently, clinical uses of escitalopram have been expanded to treat chronic pain and seasonal affective disorder [45], suggesting that the role of escitalopram is not limited to SSRI for the treatment of depressive disorders. In addition, escitalopram has adverse effects on various organs such as cardiac and nervous systems, suggesting that it might have unexpected off-target effects [67]. To explore these unidentified off-targets of escitalopram, several studies have investigated its effects on neurotransmission and ion channels by using animal model and cultured cells. Escitalopram could potentiate glutamate transmission, thereby it has faster antidepressant effect on major depressive disorders than other antidepressants [8]. Voltage-gated sodium channel and human ether-a-go-go-related gene potassium channel are directly inhibited by escitalopram [910]. However, unidentified targets of escitalopram still need to be investigated.
5-HT type 3 (5-HT3) receptor is a cation-selective ligand-gated ion channel that is gated by 5-HT. It could influence membrane depolarization and modulate neurotransmitter release at the presynaptic terminal [111213]. It is a member of the Cys-loop receptor family, which has a pentameric structure together with nicotinic acetylcholine receptors, γ-aminobutyric acid (GABA) type A receptors, and glycine receptors [1415]. 5-HT3 receptor is widely distributed in the central nervous system including postrema and nucleus tractus solitaries, hippocampus, cingulate cortex, amygdala, frontal cortex, nucleus accumbens, and dorsal horn. It is also expressed at dorsal root ganglia, colon, kidney, and immune cells [1316]. Such wide distribution of 5-HT3 receptor suggests its potential therapeutic effects on emesis, irritable bowel syndrome, schizophrenia, anxiety, substance abuse and addiction, analgesic action, and inflammatory actions [131617]. However, only antiemetic efficacy of 5-HT3 receptor antagonists is widely used clinically [1819]. For other disorders, the pathophysiologic role of 5-HT3 receptor is currently not clear enough to demonstrate the therapeutic efficacy of 5-HT3 receptor antagonist, although previous studies have suggested the possibility of its effectiveness [1820212223]. To understand the pathophysiologic role of 5-HT3 receptor in depressive disorder, past studies have tested whether 5-HT3 receptor antagonists could improve symptom of depressive disorder [24]. However, only a few antidepressants have been tested to explore its inhibitory mechanism on 5-HT3 receptor [252627]. Thus, the objective of this study was to determine the direct effect of escitalopram on 5-HT3 receptor currents using voltage-clamping technique to suggest the possible mechanism of action of escitalopram in treating depressive disorders without being limited to the function of SSRI.
METHODS
Cell culture
NCB-20 neuroblastoma cells provided by Dr. Lovinger (National Institute on Alcohol Abuse and Alcoholism, USA) have already been revealed to show 5-HT-induced current mediated by 5-HT3 receptor, making it possible to test drug effect on 5-HT3 receptor [28293031]. They were incubated in culture flasks filled with culture medium consisting of 89% Dulbecco Modified Eagle's Medium, 10% fetal bovine serum, and 1% hypoxanthine aminopterin thymidine supplement. Cells were grown in an incubator at 37℃ with 5% CO2. Cells were harvested after treatment with 0.25% trypsin-EDTA. They were then seeded to a treated culture dish at 24–48 h prior to recording. Culture medium in dish was exchanged to extracellular solution, after which cells were moved to a recording chamber 2 h before recording.
Whole cell voltage-clamp recording
Extracellular solution contained 150 mM NaCl, 2.5 mM KCl, 2.5 mM CaCl2, 0.1 mM MgCl2, 10 mM D-glucose and 10 mM N-(2-hydroxyethyl)piperazine-N′-2-ethansulfonic acid (HEPES). Its pH was adjusted to 7.4 with NaOH and its osmolality was adjusted to 340 mOsm/kg with sucrose. Intracellular solution contained 140 mM CsCl, 2 mM MgCl2, 5 mM ethylene glycol bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 10 mM HEPES. Its pH was adjusted to 7.2 with CsOH and its osmolality was adjusted to 310 mOsm/kg with sucrose. DM IRM inverted microscope (Leica, Wetzler, Germany) was used to visualize cells. Borosilicate glass capillaries (1B150-4; World Precision Instruments, Sarasota, FL, USA) were used to make recording pipettes with a horizontal micropipette puller (P-97; Sutter Instrument, Novato, CA, USA). Pipette resistance was around 2.0 MΩ when it was filled with an intracellular solution. 5-HT3 receptor currents were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA). They were filtered at 2 kHz and digitized at 10 kHz. Digidata 1322 and pClmap9 software (Molecular Devices) were used to save the recording data on a PC. Pipette capacitance was compensated after achieving giga-ohm seals with holding potential of −50 mV. Whole cell capacitance and series resistance were compensated after making whole-cell. Leak subtraction was not used in this study. All experiments were performed at room temperature (25℃). Holding potential was −50 mV except when testing current-voltage relationship.
Drug application system
An extracellular solution was continuously perfused at each side of the theta glass micropipette which were pulled from theta glass capillary tubes (Clark Borosilicate Theta; Warner Instruments, Hamden, CT, USA; 2 mm outer diameter, 1.4 mm inner diameter, 0.2 mm septum thickness) to an outer diameter of ~300 µm. After making a whole-cell configuration, cell was gently lifted with a 4-axis motorized micromanipulator (QUAD; Sutter Instrument) from the bottom of the recording chamber. Cell was moved to the side of theta glass micropipette which was flowing extracellular solution. Perfusion of extracellular or drug solutions through theta glass pipette was turn-on and -off by a perfusion valve control system (VC-8; Warner Instruments). While cell was located on the side of the extracellular solution flow at the theta glass micropipette, the agonist or escitalopram mixed agonist solution was perfused on the other side of theta glass tubing. Theta glass micropipette was mounted onto a piezo actuator (P-601 PiezoMove Linear Actuator; Physik Instrumente, Karlsruhe, Germany) which moved theta micropipette laterally in msec time resolution. Therefore, cell could be switched rapidly to the drug solution flow of theta micropipette from the extracellular solution flow. After cell was exposed to the drugs for a programmed time, it rapidly returned to the original side of the theta micropipette perfused with the extracellular solution.
Data analysis
Peak amplitudes, time constants, and decay slopes of currents were analyzed from the recorded data by using statistical tools in Clampfit 10 software (Molecular Devices). Decay slopes of currents were measured between 10% to 90% of the maximal amplitude. Each current amplitude was normalized to the peak current amplitude induced by 10 µM 5-HT. Normalized 5-HT or escitalopram concentration-peak amplitude data were fitted to the four-parameter logistic equation using Prism 8.0 (GraphPad Software, San Diego, CA, USA).
Equation (1) was used to calculate EC50.

The bottom of equation (1) was minimal response while the top was maximal response. X was the logarithm of 5-HT concentration and EC50 was the concentration of 5-HT producing a half response between bottom and top.
Equation (2) was used to calculate IC50.

The bottom of equation (2) was minimal response while the top was maximal response. X was the logarithm of escitalopram concentration and IC50 was the concentration of escitalopram resulting in a half response between bottom and top.
Interaction kinetics of escitalopram to opened 5-HT3 receptor are described on the basis of a first-order blocking scheme [32]. Apparent rate constants for association (k+1) and dissociation (k−1), and dissociation constant (Kd) were calculated from the following equations:


where τ was the time constant of binding of escitalopram on open 5-HT3 receptors analyzed by fitting decay of currents by escitalopram (for 5 sec) 3 sec after 5-HT application to the single exponential function.
Mean and SEM value were calculated using Prism 8.0. Statistical significance was set at p < 0.05. Student's t-test, ANOVA, and Tukey's multiple comparisons test were used for data analysis.
RESULTS
Inhibitory effect of escitalopram on 5-HT3 receptor-mediated currents
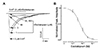
First, we evaluated concentration-dependent inhibitory effect of escitalopram on 5-HT3 receptor (i.e., 1, 3, 10, 30, and 100 µM of escitalopram was co-applied with 3 µM of 5-HT, near the EC50 value from our previous experiment [33]). Current peak amplitudes were then normalized to 5-HT3 receptor current induced by 3 µM 5-HT alone. Drugs were applied for 5 sec with 1 min interval. Fig. 1A shows superimposed representative current traces induced by application of 3 µM 5-HT alone or with various concentrations of escitalopram. As seen in Fig. 1B, 5-HT3 receptor currents were inhibited by escitalopram in a concentration-dependent manner. Using equation (2) in Materials and Methods, the IC50 value of escitalopram for 3 µM 5-HT induced currents was 5.35 ± 0.95 µM and Hill slope was −1.28 ± 0.13 (n = 10).
Effects of excitalopram on 5-HT concentration-response relationship
To test whether the inhibitory effect of escitalopram was competitive or non-competitive, we applied 5-HT at various concentrations (0.3, 1, 3, 10, 30 µM) along with escitalopram at 10 µM which was above IC50 value calculated in Fig. 1B. Fig. 2A shows representative 5-HT3 receptor current traces induced by various concentrations of 5-HT with or without escitalopram. 5-HT3 receptor currents were inhibited by escitalopram co-applied with 5-HT at all tested concentrations. Fig. 2B presents averaged 5-HT concentration-response curves with or without 10 µM escitalopram. Peak amplitudes of 5-HT3 receptor currents were decreased by escitalopram. It failed to reach the maximal even when 30 µM 5-HT was co-applied with 10 µM escitalopram. From these experiments, we calculated EC50 using equation (1) shown in the Materials and Methods. EC50 value of 5-HT alone (n = 9) and coapplied with escitalopram (n = 9) were 3.13 ± 0.11 µM and 6.36 ± 0.18 µM, respectively. Hill slope of 5-HT alone and co-applied with escitalopram were 2.58 ± 0.14 and 2.74 ± 0.14, respectively. Thus, escitalopram shifted the 5-HT concentration-response curve for 5-HT3 receptor currents to the right (p < 0.001, unpaired t-test) without significantly changing Hill coefficient (p = 0.4570, unpaired t-test). Escitalopram also significantly decreased the maximal response of 5-HT3 receptor currents evoked by 30 µM 5-HT; 103.0 ± 1.7% of 10 µM 5-HT induced current for 30 µM 5-HT alone and 87.8 ± 2.1% for 30 µM 5-HT co-applied with escitalopram (p < 0.001, unpaired t-test). These results suggest that escitalopram can inhibit 5-HT3 receptor function in a non-competitive manner because escitalopram increases EC50 of 5-HT on the 5-HT3 receptor currents concomitant with a decrease of maximal effect.
Voltage independency of escitalopram on 5-HT3 receptor inhibition
We next tested voltage dependency of the inhibitory effect of escitalopram on 5-HT3 receptor currents. We applied 3 µM 5-HT with or without 10 µM escitalopram to cells at holding potentials of −50, −30, −10, 0, 10, and 30 mV. Left side of Fig. 3A shows superimposed current traces of 3 µM 5-HT at various holding potentials. Right side of Fig. 3A shows 3 µM 5-HT induced currents co-applied with 10 µM escitalopram. Peak amplitude of each current was normalized to the peak amplitude of 3 µM 5-HT. Averaged results are presented in Fig. 3B. Peak currents were decreased at all tested holding potentials ranging from −50 mV to 30 mV. Reversal potential of 5-HT induced currents was 7.53 ± 0.53 mV (n = 9) by 5-HT alone and 8.43 ± 0.64 mV (n = 9) by 5-HT co-applied with escitalopram, showing no significant difference in the reversal potentials between the two (p = 0.2893, unpaired t-test). The ratio of peak amplitude between currents with or without escitalopram (I5-HT+ES/I5-HT) was not changed in any holding potential (Fig. 3C, one-way ANOVA, F = 0.7624, p = 0.5560). These results (i.e., reversal potential was not changed and inhibitory ratio was not significantly different among various holding potentials) suggested that the inhibitory effect of escitalopram was voltage independent.
Differences in inhibition between pre-application and co-application of escitalopram with 5-HT
In order to study whether escitalopram could bind to open form or closed form of 5-HT3 receptor ion channel, we tested effects of different application modes of escitalopram (10 µM) on 3 µM 5-HT induced currents and then compared its inhibitory effect between application modes. Fig. 4A–D show representative current traces of 3 µM 5-HT induced currents, absence of escitalopram (Fig. 4A), co-application of escitalopram (Fig. 4B), continuous application of escitalopram from 1 min prior to 5-HT application (Fig. 4C), and pre- and post-application without coapplication of escitalopram (Fig. 4D). Peak amplitudes of each application method of escitalopram was normalized to 3 µM 5-HT response. Averaged data are shown in Fig. 4E. Escitalopram inhibited peak amplitudes of 5-HT currents at each application mode. However, escitalopram exerted larger inhibition in continuous application mode (n = 10, 21.6 ± 2.7% of 3 µM 5-HT induced currents) than co-application without pretreatment (n = 10, 34.5 ± 2.8% of 3 µM 5-HT induced currents) (p < 0.001, AVOVA and Tukey's multiple comparison test). The inhibitory effect of escitalopram in pretreatment only was much smaller than continuous application and co-application modes (n = 10, 65.4 ± 3.7% of 3 µM 5-HT induced currents, p < 0.001, AVOVA and Tukey's multiple comparison test). These results suggest that escitalopram can inhibit 5-HT3 receptor by binding to both open and closed state of 5-HT3 receptor ion channel, although it preferentially acts on the open state of 5-HT3 receptor ion channel. This was because 5-HT3 receptor currents still remained even in continuous application of escitalopram (Fig. 4C). In addition, inhibitory effect by pretreatment only was not profound (Fig. 4D).
Interaction kinetics of escitalopram on opened 5-HT3 receptor
After 3 sec application of 3 µM 5-HT, escitalopram at various concentrations (1, 3, 10, 30, and 100 µM) was co-applied with 5-HT for 5 sec to investigate association and dissociation kinetics of escitalopram binding on open state of 5-HT3 receptor. Superimposed representative traces are shown in Fig. 5A. Escitalopram accelerated current decays during co-application. Currents reappeared upon discontinuation of escitalopram co-application at concentration higher than 10 µM. Association time constants for escitalopram on open 5-HT3 receptors were obtained by single exponential function fitted to current decays during escitalopram co-application. Dissociation time constants for escitalopram were obtained by single exponential function fitted to current risings upon termination of escitalopram co-application. Escitalopram decreased association time constants in a concentrationdependent manner from 1 to 100 µM. Although dissociation time constants were measurable in concentration range of 10–100 µM escitalopram, there were no significant changes in dissociation time constants upon treatment with different escitalopram concentrations (Fig. 5B). Fig. 5C shows a linear relationship of reverse values of association time constants depending on the concentration of escitalopram obtained from equation (3) as shown in the Methods. The slope of this linear function (meaning association rate constant (k+1) of escitalopram on open 5-HT3 receptors) was 0.056 µM−1sec−1. Y intercept value meaning dissociation rate constant (k−1) was 0.279 sec−1. From these association and dissociation rate constants, the calculated Kd value on open 5-HT3 receptors using equation (4) shown in the Methods was 4.99 µM. This value was very close to the IC50 of escitalopram on 5-HT3 receptor currents as shown in Fig. 1.
Effects of escitalopram on receptor desensitization kinetics
To study change of desensitization process of 5-HT3 receptor caused by escitalopram, we applied 10 µM 5-HT for 10 sec to induce strong desensitization and analyzed decay slopes of currents during application of 5-HT alone or with 10 µM escitalopram. As shown in Fig. 6A, current decay during 5-HT application was accelerated by escitalopram. Averaged decay slopes were increased from 0.1114 ± 0.0170 pA/msec to 0.3027 ± 0.0458 pA/msec by escitalopram (Fig. 6B, n = 10, p < 0.05, paired t-test). This indicated that escitalopram forced the open 5-HT3 receptor channel to close or desensitization, supporting the mechanism of open channel blocking. Effects of escitalopram on the time course of recovery from desensitization were studied using two pulses of 5-HT application in variable inter-pulse intervals (1, 5, 10, 30, 60 sec). Peak amplitude of the second pulse was then normalized to the first peak amplitude (paired-pulse ratio) and plotted on inter-pulse intervals. Fig. 6C and D show representative superimposed current traces evoked by two pulses of 10 µM 5-HT for 5 sec in the interpulse intervals of 1, 5, 10, 30, 60 sec with (Fig. 6D) or without (Fig. 6C) 10 µM escitalopram. A single exponential function was fitted to the 5-HT3 receptor desensitization recovery time course with a time constant of 8.80 ± 0.63 sec (n = 9) for 5-HT alone and 8.94 ± 0.70 sec (n = 9) for 5-HT co-applied with escitalopram (Fig. 6E, p = 0.8826, unpaired t-test). These results suggested that escitaolpram did not change 5-HT3 receptor desensitization recovery time course.
DISCUSSION
In this study, escitalopram reduced peak amplitude of 5-HT3 receptor current in a concentration-dependent manner. Current induced by 5-HT was almost completely blocked by escitaolpram at concentration higher than 30 µM. Escitalopram functioned as a non-competitive antagonist. Thus, escitalopram not likely bound to agonist binding sites. The inhibitory effect of escitalopram showed voltage-independency. It was more effective in the co-application mode than pre-application alone. It also accelerated the 5-HT3 receptor desensitization. The effectiveness of preapplication represents that antagonist can act as a closed channel blocker and that of co-application represents that antagonist can act as an open channel blocker [34]. Open channel antagonism is associated with channel pore blocking when it shows voltage-dependency [3435]. In our study, escitalopram showed an inhibitory effect in both pre-application mode and co-application mode, although the co-application mode was more effective than pre-application alone. This means that escitalopram prefers open state inhibition than close state inhibition. The voltage-independency from our results suggests that escitalopram is not a pore blocker. Taken together, these results indicate that escitalopram does not share the binding site with 5-HT and that it does not bind at the channel pore. Thus, allosteric site could be the most potential candidate binding site for escitalopram. Furthermore, open channel inhibition of escitalopram during co-application could be explained by allosteric modulation because escitalopram could bind to receptor independently from binding to 5-HT. Allosteric binding might also happen at close state of 5-HT3 receptor. Thus, escitalopram showed inhibitory effect in both pre-treatment mode and co-application mode. These characteristics were consistent with previous studies revealing that non-competitive inhibition with open channel block was mediated by allosteric modulation [3637]. After binding of escitalopram at allosteric site, binding affinity of 5-HT on 5-HT3 receptor could be changed, thus inhibiting the channel to open or accelerating the receptor to be desensitized. The calculated Kd value in Fig. 5C was close to the IC50 obtained from peak amplitude analysis shown in Fig. 1B. This finding could confirm these interpretations.
In addition, escitalopram showed fast association and dissociation to open 5-HT3 receptor because currents inhibited rapidly by escitalopram and rapidly re-occurred upon termination of escitalopram co-application concentration-dependently. This finding support the faster-acting characteristics of escitalopram for treating patients with major depressive disorder than other SSRIs [38]. Because 5-HT3 receptors are located in presynaptic terminals to modulate release of neurotransmitters including 5-HT [3940], an inhibitory effect of escitalopram on presynaptic 5-HT3 receptor could modulate the release of 5-HT from synaptic terminals. This effect could be additive to secondary changes of synaptic transmission, such as desensitization of presynaptic autoreceptors and inhibition of neurotransmitter synthesis [41] following a primary mechanism of escitalopram (i.e., serotonin reuptake blocking). This additive effect could contribute to pharmacologic characteristics of escitalopram, such as faster-acting than other SSRIs and withdrawal symptoms.
In conclusion, we demonstrated an inhibitory effect of escitalopram on 5-HT3 receptor by allosteric modulation. Escitalopram acted through the mechanism of open channel blocking. It underwent fast association and dissociation, reflecting its fast effectiveness and possibility of its withdrawal symptom. These results suggest that the therapeutic effect of escitalopram could be due to direct inhibition of 5-HT3 receptor function in addition to primary serotonin reuptake blocking.




 XML Download
XML Download