

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Inflammation is a self-protection response against pathogens, chemicals, or mechanical injuries. However, hyperinflammation can cause adverse effects in tissues and organs, which ultimately leads to chronic inflammatory diseases [1]. A number of studies have demonstrated that macrophages are the primary inflammatory response cells [23]. Their activation causes the overproduction of inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, nitric oxide (NO), and prostaglandin (PG)E2 [4].
Nuclear factor-κB (NF-κB) serves as a key regulator of inflammation and innate immune response by control of the expressions of numerous inflammatory genes [56]. For example, binding of NF-κB to a unique sequence (NF-κBd) in the inducible nitric oxide synthase (iNOS) promoter is necessary for lipopolysaccharide (LPS)-induced iNOS expression [7]. Similarly, the 5'-flanking region of murine cyclooxygenase-2 (COX-2) gene contains two NF-κB binding consensus sequences [8]. LPS stimulation of macrophages is known to activate mitogenactivated protein kinases (MAPKs) such as extracellular signalregulated kinase (ERK)-1/2, p38 MAPK, and c-Jun N-terminal kinase (JNK) [9]. Hence, NF-κB and MAPKs pathways are potentially important targets for regulating macrophage associated inflammation.
Rhodanthpyrone (Rho) A and B are the primary active compounds in the plant Gentiana rhodantha, a traditional Chinese medicine for the treatment of inflammation [10]. However, to the best of our knowledge, their biological properties have never been reported. Therefore, in the present study, we elucidate the anti-inflammatory effects and mechanisms of action of RhoA and RhoB, using LPS-stimulated macrophages.
METHODS
Cell culture and reagents
The murine macrophage cell line RAW 264.7 and 3T3-L1 fibroblasts were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were maintained at 37℃ in a humidified, 5% CO2 atmosphere, and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, 50 U/ml penicillin, 50 µg/ml streptomycin, and 2.5 µg/ml of amphotericin B. 3T3-L1 cells were induced to differentiate and the adipocyte conditioned medium (CM) was prepared as described elsewhere [1112]. RhoA and RhoB were prepared as previously described [13]. All reagents used in this study were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise noted.
Isolation of peritoneal macrophages
C57BL/6J mice were intraperitoneally injected with 2 ml of sterile thioglycollate medium from BD Biosciences (San Jose, CA, USA). Macrophages were collected three days later by peritoneal lavage with cold DMEM. The recovered peritoneal fluid was washed twice with PBS by centrifugation. The cells were then resuspended in DMEM with 10% fetal bovine serum and incubated for 3 h at 37℃ with 5% CO2. Non-adherent cells were removed.
Cell viability
The viability of RAW 264.7 cells was determined by analyzing the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan. In brief, after 24 h of incubation, cells in 96-well plates were washed twice with PBS, and MTT (0.5 mg/ml in PBS) was added to each well. Cells were then incubated at 37℃ for 3 h, and DMSO (100 µl) was added to dissolve the formazan crystals. Absorbance was measured at 570 nm using the Spectra MAX PLUS spectrophotometer from Molecular Devices (Sunnyvale, CA, USA).
NO measurement
NO levels were estimated by measuring nitrite levels (a stable metabolite of NO) using the Griess reagent, as previously described [14]. Briefly, RAW 264.7 cells were pretreated with different concentrations of RhoA and RhoB for 1 h prior to the addition of LPS. After 24 h, 100 µl aliquots of culture supernatants were mixed with an equal volume of a modified Griess reagent at room temperature for 20 min, and the absorbance at 540 nm was measured using a spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The modified Griess reagent was a 1:1 mixture of 1% sulfanilamide in 30% acetic acid, and 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride in 60% acetic acid.
Western blot analysis
Cell lysates containing 20 µg of total protein were separated using 10% SDS-PAGE and transferred to nitrocellulose membranes. After blocking with 5% skim milk, the blot was probed with primary antibodies against iNOS (BD610333) from BD Biosciences (Palo Alto, CA, USA); COX-2 (sc-1745), IκBα (sc-371), and actin (sc-1616) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); p-p65 (Ser311, ab51059) from Abcam (Cambridge, MA, USA); and p-JNK (#92515), p-p38 (#4631), p-IKKα/β (#2697), and p-ERK (#9101) from Cell Signaling Technology (Beverly, MA, USA). Immunoreactive bands were detected using a Las-4000 imager from GE Healthcare Life Science (Pittsburgh, PA, USA).
RNA isolation and quantitative real-time RT-PCR (qPCR)
RNA was extracted from cells using TRIzol reagent from Invitrogen (Carlsbad, CA, USA). Total RNA (2 mg) was treated with RNase-free DNase from Invitrogen, and first-strand cDNA was generated using a random hexamer primer, using a first-strand cDNA synthesis kit from Promega (Madison, WI, USA). Specific primers were designed using qPrimerDepot (https://pga.mgh.harvard.edu/primerbank/, Table 1). qPCR reactions consisted of 10 ng reverse-transcribed total RNA, 200 nM forward and reverse primers, and 2 × PCR master mix, with a final volume of 10 µl. Reactions were carried out in 384-well plates using an ABI Prism 7900HT Sequence Detection System from Applied Biosystems (Foster City, CA, USA). The mRNA expression of all genes tested was normalized to the Rps3 expression.
Enzyme-linked immunosorbent assays (ELISA)
RAW 264.7 cells in 12-well plates were pretreated with either RhoA or RhoB, and treated with LPS for 24 h. Protein levels of C-C Motif Chemokine Ligand 2 (CCL2, also known as monocyte chemoattractant protein-1, MCP-1) from Peprotech (Rocky Hill, NJ, USA) and TNF-α from Enzo Life Sciences (Plymouth Meeting, PA, USA) in cell-free supernatants were analyzed using ELISA kits.
In vitro chemotaxis assay
RAW 264.7 cell migration assays were performed in transwell migration assay chambers (BD Life Sciences, Franklin Lakes, NJ, USA) by adding adipocyte-derived CM to the lower chamber. Raw 264.7 cells incubated with vehicle, RhoA or RhoB for 3 h were seeded onto transwell and the migrated cells towards adipocyte CM were counted after 3 h exposure.
Transient transfection and luciferase reporter assay
RAW 264.7 cells were seeded on 6-well plates. For the reporter gene assay, cells were transfected with 2 µg/ml NF-κB luciferase from Stratagene (La Jolla, CA, USA) and 1 µg/ml pRL-TK from Promega using Lipofectamine 2000 from Invitrogen (Carlsbad, CA, USA). Cells were pretreated with RhoA or RhoB for 1 h, and then treated with 10 ng/ml LPS for 24 h. After cell lysis, luciferase activity was measured using a Dual Luciferase Reporter assay kit from Promega.
RESULTS
RhoA and RhoB inhibit LPS-induced iNOS and COX-2 expression in macrophages
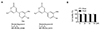
The chemical structures of RhoA and RhoB are shown in Fig. 1A. We first determined the non-cytotoxic concentration range of RhoA and RhoB in RAW 264.7 cells. Neither RhoA nor RhoB (up to 80 µM) affected cell viability (Fig. 1B). Therefore, concentrations less than or equal to 80 µM were selected for further examination.
Since NO has been implicated in LPS-mediated inflammation, we analyzed the effect of RhoA or RhoB on NO production in LPS-stimulated RAW 264.7 cells. The cells were pretreated with various concentrations of RhoA or RhoB for 1 h prior to LPS (10 ng/ml) stimulation and further incubated for 24 h. Supernatants of LPS stimulated cells had significantly increased nitrite (a stable oxidized product of NO) levels, compared with the controls. Pretreatment with either RhoA or RhoB inhibited LPS-induced NO production in a concentration-dependent manner, with maximum inhibition at 80 µM for RhoA and 40 µM for RhoB (Fig. 2A). To examine if RhoA and RhoB inhibit NO production via suppression of iNOS (Nos2) gene expression, the changes in iNOS mRNA and protein levels in RAW 264.7 cells were investigated by qPCR and Western blotting, respectively. LPS treatment increased iNOS protein levels as early as 5 h and plateaued at 10 h after the treatment, while RhoA or RhoB pretreatment abrogated the increases in iNOS levels at all-time points (Fig. 2B). Additionally, RhoA and RhoB inhibited iNOS protein expression in LPS-stimulated RAW 264.7 cells in a concentration-dependent manner (Fig. 2C). The transcript levels of iNOS after 6 h exposure of RhoA or RhoB were also suppressed (Fig. 2D). Moreover, 24 h treatment of RhoA or RhoB inhibited iNOS expression in thioglycollate-elicited mouse peritoneal macrophages in a dose-dependent manner (Fig. 2E). Results for COX-2 expression were the same (Fig. 2B–E).
RhoA and RhoB suppress LPS-induced production of pro-inflammatory mediators in RAW 264.7 cells
Effects of RhoA and RhoB on proinflammatory cytokines/chemokine mRNA expression and protein production in RAW 264.7 cells exposed to LPS were investigated using qPCR and ELISA analyses. qPCR analysis revealed that LPS treatment significantly increased the mRNA levels of CCL2, TNF-a, and IL-6, and that these increments were suppressed by pretreatment with RhoA or RhoB (Fig. 3A). Subsequent quantitative analysis using ELISA confirmed the reduced production of CCL2 and TNF-α; LPS treatment in the control group significantly increased CCL2 and TNF-α protein levels by 12.0 ± 2.9 and 118.7 ± 7.0 folds, respectively. Pretreatment with 40 µM RhoA in the control group reduced LPS-induced CCL2 and TNF-α secretion by 4.7 ± 0.8 and 74.6 ± 2.3 folds, respectively, compared with the LPS group (Fig. 3B). RhoB treatment also significantly inhibited CCL2 and TNF-α levels (Fig. 3B). Moreover, treatment with RhoA and RhoB diminished the migration of RAW 264.7 cells exposed to CM derived from adipocytes (Fig. 3C), indicating the inhibitory effect of RhoA and RhoB on macrophage chemotaxis.
RhoA and RhoB suppress the LPS-stimulated NF-κB signaling pathway in RAW 264.7 cells
Since NF-κB activation is crucial for regulating iNOS, COX2, and proinflammatory cytokines expression, we analyzed the NF-κB signaling pathway in LPSstimulated RAW 264.7 cells, using western blotting. Results revealed that LPS treatment increased the phosphorylation levels of inhibitory κB kinase (IKK) and p65, and decreased inhibitory κB kinase α (IκBα), which were successfully attenuated by either RhoA or RhoB pretreatment (Fig. 4A). Suppression of NF-κB by RhoA or RhoB was further confirmed using the NF-κB luciferase assay (Fig. 4B). These results indicate that RhoA and RhoB inhibit IκBα degradation, thus, preventing the activation of NF-κB.
Along with the suppression of NF-κB, the effects of RhoA and RhoB on MAPK signaling were also investigated. As shown in Fig. 4C, LPS alone significantly increased phosphorylation of JNK, ERK, and p38 MAPK 30 min after stimulation. However, in contrast to their effect on the NF-κB pathway, RhoA and RhoB did not affect the phosphorylation status of three MAPK pathways, i.e. ERK, JNK and p38 MAPK, in RAW 264.7 cells (Fig. 4C). Thus, we further verified the role of NF-κB signaling in the anti-inflammatory effects of RhoA and RhoB. Treatment with an NF-κB inhibitor blocked LPS-induced iNOS and COX-2 expressions, as did RhoA and RhoB (Fig. 4D). In addition, the inhibitory effect of RhoA and RhoB was attenuated in the condition of NF-κB inhibition (Fig. 4D). Collectively, these data suppose that RhoA and RhoB inhibit inflammatory responses in macrophages at least in part by inhibiting NF-κB pathway.
DISCUSSION
Macrophage-associated inflammation contributes to the pathogenesis of diverse diseases. Activated macrophages produces numerous pro-inflammatory mediators, which are crucial for the progression of acute and chronic liver diseases including viral hepatitis, non-alcoholic or alcoholic steatohepatitis, and metabolic disorders such as obesity and diabetes [2315]. The main finding of the current study is the discovery of RhoA and RhoB as novel candidate compounds having the anti-inflammatory activities. Several cell models using macrophages revealed that noncytotoxic concentration (mainly, 40 to 80 µM) of RhoA and RhoB significantly reduced the expression of inflammatory genes, as well as the secretion of various pro-inflammatory cytokines/chemokine (i.e., Ccl2, Tnfa, and Il6) in LPS-stimulated condition. The anti-inflammatory effects of RhoA and RhoB on iNOS and COX2 expressions were also verified in primary murine macrophages. They also decreased NO production, being consistent with the repression of iNOS. The data support the possible protective roles of these compounds in inflammatory diseases.
Adipocytes serve as one of the sources of production of various pro-inflammatory cytokines (e.g., TNF-a, and IL-6) and chemokines (e.g., CCL2) upon metabolic stress [16]. Therefore, macrophage infiltration in adipose tissues is critical for obesity-linked insulin resistance [1718]. Another important finding of the present study is that RhoA and RhoB inhibit macrophage chemotaxis in response to adipocyte-derived CM, possibly raising the value of the agents with beneficial effects on metabolic diseases with inflammation.
As the molecular mechanisms of the anti-inflammatory actions of RhoA and RhoB, we found that the agents suppress the NF-κB signaling pathway. More specifically, treatment of RhoA and RhoB before LPS stimulation resulted in decreased p-IKK/IKK and p-p65/p65, and increased IκBα, which are hallmarks for NF-κB suppression. As a consequence, RhoA and RhoB also markedly decreased the transcriptional activities of NF-κB, as shown in luciferase assays. Furthermore, the inhibition of NF-κB activity diminished the ability of RhoA and RhoB to suppress the inflammatory gene expressions under LPS-stimulated condition, supporting the possible role of NF-κB inhibition in anti-inflammatory actions of RhoA and RhoB. MAPKs have been known to be involved the pathogenesis of LPS-induced inflammation [919], but the regulation of MAPK showed pro-inflammatory and anti-inflammatory responses at different time points, indicating the complex and dynamic regulation of the signaling pathway. In addition, the effects of MAPKs on the activation of NF-κB seemed unclear [20]. In our experimental condition, RhoA and RhoB did not affect canonical three MAPK signaling pathways, suggesting that the inhibition of NF-κB signaling is independent of MAPKs pathway. Activation of NF-κB-mediated inflammatory events is involved in the initiation and progression of diverse inflammatory diseases. TNF family cytokines also increase the activity of NF-κB [21], which may result in vicious cycle for the activation of inflammation. In this regard, controlling the NF-κB signaling pathway is a promising therapeutic strategy for the prevention and treatment of inflammation. Thus, the in vivo anti-inflammatory efficacy of RhoA and RhoB should be further explored.
In conclusion, our findings suppose that RhoA and RhoB have the anti-inflammatory properties, which might be mediated at least in part by suppressing the NF-κB signaling pathway in macrophages. It also may provide a novel approach to overcome inflammation-associated diseases.




 XML Download
XML Download