

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Neurofibromatosis type 1 (NF1), also known as Von Recklinghausen's disease, is an autosomal dominant hereditary disorder, with an estimated incidence of 1 out of 3,000 births.1 The presentation of NF1 is characterized by cutaneous neurofibromas, café-au-lait spots, axillary and inguinal freckling, and Lisch nodules. The pathogenesis of NF1 has been suggested to be an alteration of the NF-1 gene, which is located on chromosome 17q 11.2.2 Because NF-1 encodes neurofibromin, which controls cellular proliferation, a mutation of NF-1 causes a loss of tumor suppressor function, which leads to the development of benign and malignant tumors in many sites as well as cutaneous disorders.2
Previous studies reported a 5% to 25% frequency of intra-abdominal tumors of NF1, and it may be affected by the manifes tation of symptoms.3 According to the histopathology, gastrointestinal (GI) tract involvement in NF1 can be categorized as follows:4 neurogenic neoplasm (neurofibroma, plexiform neurofibroma, and ganglioneuroma), neuroendocrine tumors (NETs; carcinoid, pheochromocytoma, and paraganglioma), non-neurogenic mesenchymal tumors (gastrointestinal stromal tumor [GIST], leiomyoma, and leiomyosarcoma), embryonal tumors (rhabdomyosarcoma, neuroblastoma, and Wilm's tumor), and miscellaneous tumors (GI adenocarcinoma). GISTs are the most common GI tract neoplasms associated with NF1.5
Despite the many case reports on the coexistence of GIST or NET with NF1, the synchronous manifestation of GIST and NET in the background of NF1 is extremely rare. This paper reports three cases of synchronous ampullary NET and GIST in association with NF1 that were treated with surgical intervention and long-term follow-up.
CASE REPORT
Case 1
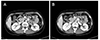
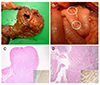
A 36-year-old woman was referred for the evaluation and treatment of a periampullary mass found on esophagogastroduodenoscopy (EGD) performed at a local clinic with the chief complaint of postprandial epigastric pain (Fig. 1). A physical examination revealed café-au-lait spots and multiple cutaneous neurofibromas over the trunk, and she had a familial history of similar cutaneous lesions in the first-degree relatives (father, sister, and daughter). She met the diagnostic criteria of NF1.6 Laboratory analysis showed no abnormal findings, including the levels of serum CEA and CA 19-9. Abdominal CT and MRI revealed a 1.4 cm well-marginated lesion in the periampullary duodenum showing peripheral enhancement in the arterial phase and homogeneous enhancement in the delayed phase as well as another 1.6×1.3 cm mass in the duodenojejunal junction with intense homogeneous enhancement (Fig. 2). Several round lymph nodes, 1.2 cm in size, at the maximum were found around the celiac axis and mesenteric root. An endoscopic biopsy performed at the local clinic revealed a well-differentiated NET. The patient underwent a pylorous-preserving pancreatoduodenectomy for the purpose of a wide regional resection and lymph node dissection. Intraoperatively, several firm and enlarged lymph nodes were found and dissected widely. The surgical specimen (Fig. 3) contained an approximately 1.5 cm-sized luminal protruding ampullary mass and another 1.6 cm sized palpable submucosal mass at the proximal jejunum. Microscopically, the jejunal submucosal mass was GIST and its mitotic figures were not observed (Fig. 4A). Immunohistochemical staining was positive for c-Kit, CD34 and synaptophysin, and CD56. The S100 stain was negative. A histopathological examination of the periampullary mass revealed a well-differentiated neuroendocrine carcinoma (Fig. 4B) without lymphovascular or perineural invasion. All the resection margins were free of invasion, but nine out of 17 lymph nodes harvested from the celiac axis and root of the superior mesenteric artery and vein were positive for invasion. The patient's postoperative recovery was uneventful and she was discharged 18 days after surgery. Three months after surgery, PET-CT revealed a focal hypermetabolic lesion at the upper central abdomen around the greater omentum. This suggested a postoperative change in the greater omentum, but the possibility of early recurrence could not be excluded. Therefore, adjuvant chemotherapy, consisting of etoposide and cisplatin over a period of 9 weeks (3 cycles), was initiated. She remains well and has been in continuous remission since her surgery in June, 2010.
Case 2
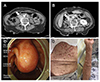
A 55-year-old man with known NF1 was admitted to the authors' institute for a duodenal mass that was detected incidentally by EGD performed for a health-care survey. Abdomen CT and MRI revealed an approximately 2 cm sized, well-defined enhancing mass, in the medial wall of the duodenal 2nd portion. No lymphadenopathy or local infiltrations were found. The results of the routine laboratory tests were all within the normal limits. EGD showed a protruding mass with superficial erosion at the site of the suprapapillar area. A biopsy of the mass was done and the reported histopathology was chronic duodenitis. A pancreatoduodenectomy was performed under the suspicion of a malignant tumor depending on its configuration. Intraoperatively, several small nodules were palpated in the duodenal 4th portion and jejunum, in addition to the known periampullary mass. The surgical specimen showed a 3×3 cm-sized hard mass over the ampula of Vater (Fig. 5A) as well as multiple nodules in the submucosal layer of the duodenum and proximal jejunum (Fig. 5B). Histopathological analysis showed that the ampullary mass was a well-differentiated neuroendocrine carcinoma invading the pancreas that was positive for lymph node invasion (Fig. 5D). Among the 11 lymph nodes harvested, four nodes placed at the head of the pancreas were positive. In addition, multiple submucosal masses located at the duodenum and jejunum were found to be GIST with a low risk (Fig. 5C). All resection margins were negative for tumor cell invasion. Postoperatively, the patient recovered without complications, and was discharged at postoperative day 19. After surgery, the patient has had an ordinary quality of life and a regular follow-up using an abdominal CT scan has not shown any recurrence of the tumor for 10 years after surgery.
Case 3
An 80-year-old woman visited the authors' hospital for an evaluation of a dilatation of the bile ducts detected by abdomen ultrasonography performed at a local clinic. Her first symptoms were general weakness and weight loss of 3 kg over a one-month period. A physical examination revealed multiple neurofibromas over the face, upper extremities, and trunk (Fig. 6D); her son had similar skin lesions. She was referred to the department of dermatology under the diagnosis of neurofibromatosis and underwent a skin biopsy, which confirmed NF1. Laboratory examinations showed that all the data were within the normal range, including the serum bilirubin level. Abdominal CT scans were obtained to exclude any neoplasm or lithiasis of the biliary tract. A 1.5 cm weakly enhanced mass was found at the ampulla of Vater and marked dilatation of the bile duct and pancreatic duct were observed (Fig. 6A). In addition, another 1.7 cm-sized mass was found between the pancreatic head and posterior to the duodenal 2nd portion (Fig. 6B). EGD showed a prominent papilla of Vater with ulceration (Fig. 6C). A biopsy was performed and histopathologic analysis revealed a NET, grade 1. Initially, a pancreatoduodenectomy was planned, but the patient preferred a less invasive procedure to a radical operation because of concerns of surgical complications. Transduodenal ampullectomy and separate tumorectomy of the retroduodenal area were performed. A histopathology evaluation revealed the ampullary mass to be well-differentiated NET, grade 1, without neurovascular or lymphatic invasion. Tumor-free margins were identified. The retroduodenal mass was identified as GIST with very low risk. The patient's postoperative course was uneventful and she was discharged after 14 days. Thirty-two months after surgery, there has been no evidence of tumor recurrence.
DISCUSSION
NF1 is a tumor syndrome caused by a mutation in the NF-1 gene,1 and synchronous GI tract lesions are relatively common in NF1. On the other hand, the occurrence of GI lesions in NF1 patients is mostly clinically occult.4 Clinical manifestations are variable and depend on the location and extent of mucosal involvement. The pathogenesis of the occurrence of variable kinds of neoplasms in association with NF1 has not been investigated clearly.
Salvi et al.7 pooled 252 GISTs detected in 126 NF1 patients and documented comparisons between GISTs presenting in NF1 patients and sporadic GISTs. They reported that GISTs occur in younger patients (mean age, 52.8 years vs. 61.4 years in sporadic GISTs), are located distally (jejunum 39.2%, ileus 30.6% and duodenum 19.8%), rare in the stomach (5.4%), multiple in 35.3%, smaller in size with a low mitotic rate, and follow a benign clinical course. They are usually clinically asymptomatic and are detected incidentally. Most of them were classified as low risk (64.9%)7 because of the lack of GIST-specific mutations (c-Kit and platelet derived growth factor receptor alpha). Therefore, they seldom respond to treatment with imatinib (a tyrosine kinase inhibitor).37 This review is consistent with the present three cases in terms of the location and asymptomatic presentation. In all three cases, GISTs were found at the jejunum and duodenum. Two of the patients presented with symptoms of pain and weight loss, which were believed to have originated not from GIST but from NET of the ampulla.
Relles et al.8 reviewed and analyzed 76 cases of periampullary and duodenal neoplasms in NF1; 92% of neoplasms presented with symptoms. Of these, 37% had pain, 32% had weight loss, 28% had jaundice, 22% had anemia, and 20% showed GI bleeding.8 In these cases, two patients presented with symptoms of postprandial epigastric pain and weight loss; the other patient was asymptomatic. Regarding the tumor location, 60% were in the duodenum and 31% were in the ampulla of Vater.9 An analysis of the type of synchronous periampullary tumors associated with NF1 revealed 40%, 34%, 8%, and 6% to be somatostatinoma, GIST, adenocarcinoma, and carcinoid, respectively.8
NETs have been reported only in 1–6% of NF1 patients, with the most common site being the periampullary region.89 Furthermore, ampullary NET in NF1 is rare. Ampullary NETs are detected at a younger age and are smaller due to the earlier presentation of symptoms than in extra-ampullary NETs.10 The symptoms are jaundice (60%) and non-specific abdominal pain (40%).10
Approximately 50% of periampullary NETs have lymph node metastasis.11 The ampulla of Vater is rich in lympho-vascular structures, where even small tumors can spread, which leads to lymph node metastases.12 More than half of the ampullary NET below 2 cm was found to be accompanied by lymph node metastases; metastases were identified in tumors, in which 66%, 50%, and 46% were <1 cm, 1–2 cm, and >2 cm in size, respectively.13 When lymph node metastases are suspected, a radical resection, such as pancreatoduodenectomy with lymph node dissection, is the preferred treatment modality for ampullary NETs and ampullary neuroendocrine carcinomas.9 This is consistent with the present cases in that the two cases that underwent lymph node dissection revealed lymph node metastasis. An endoscopic ampullectomy could be an alternative when the tumor is confined to the mucosal layer and with no vascular and lymphatic invasion. Transduodenal ampullectomy has been suggested to be a feasible option for relatively small and suspected submucosal invasion. In the present cases, one patient underwent a transduodenal ampullectomy. Nevertheless, these less invasive procedures have the risk of incomplete tumor removal because the preoperative imaging study and endoscopic biopsy are often inaccurate regarding lymph node involvement and depth of invasion. When a lymph node metastasis is confirmed postoperatively as synchronous tumors in the background of NF1, optimal treatments after a surgical resection, including a chemotherapy regimen have not been established. On the other hand, one study reported the effectiveness of combined chemotherapy with cisplatin and etoposide for the treatment of extrapulmonary neuroendocrine carcinoma.14 Case 1 underwent treatment with etoposide and cisplatin as adjuvant chemotherapy because PET-CT could not exclude early recurrence.
In conclusion, synchronous GIST and NET in the background of NF1 is extremely rare, but the possible coexistence of combined tumors is relatively high in NF1 patients compared to the general population. Furthermore, both NETs and GISTs occurring in NF1 patients tend to be smaller in size than in the general population. Therefore, close attention must be paid to identifying the coexistence of other neoplasms, when NF1 patients present with vague abdominal discomfort.



 XML Download
XML Download