

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Newborn screening (NBS) for inborn errors of metabolism is a worldwide public health program implemented to screen asymptomatic newborns for rare inherited diseases for which early treatment results in significant reductions in morbidity and mortality. NBS methods have evolved in recent years, especially with the development of tandem mass spectroscopy (MS/MS), which now facilitates the identification of many disorders in parallel using a single assay.12 In Korea, NBS for over 50 diseases has been supported free of cost by a national program since November 2018.
However, current NBS programs show high rates of false-positive results (FPRs). In addition, tests performed on sick, premature, and/or low birth weight infants that require hospitalization in a neonatal intensive care unit (NICU) result in higher FPRs, requiring repeated NBS and additional follow-up tests, which can significantly delay diagnosis and treatment of sick neonates.34 Thus, there is a need for faster intervention and special planning for the neonate population to make more accurate and timely diagnoses. However, few studies have addressed the best ways to reduce the FPR of conventional NBS in the NICU. The Clinical and Laboratory Standards Institute has recommended that infants suspected to have a metabolic disorder should undergo a follow-up NBS test to be performed only after ceasing total parenteral nutrition (TPN) or medical therapies initiated owing to an abnormal NBS. However, rapid diagnosis using conventional NBS methods for early intervention remains a challenge in stressed neonates.56 The recent advent of next-generation sequencing (NGS) technology has enabled more effective diagnosis of monogenic disorders for suspected specific diseases in newborns by using phenotype-based NGS panels or whole exome sequencing.4789
However, the main goal of NBS is to screen all neonates and allow for the early detection of affected infants to prevent serious clinical concerns prior to their discharge from the hospital. Inborn errors of metabolic disorders are rare genetic disorders, some of which cannot be identified by tandem mass spectrometry. Therefore, few studies have aimed to determine whether molecular diagnostics can be used to replace NBS: the Babyseq Project supported by Newborn sequencing in Genomic Medicine and Public Health is the most representative study, with 127 healthy newborns and 32 newborns admitted to NICU who were not preselected for specific diseases. Replacing NBS with molecular screening remains a challenge owing to variant analysis, interpretation, and reporting for adult-onset disorders from genomic data.1011 In this study, we describe the useful implementation of a targeted NGS panel including childhood-actionable genes to complement the problem of delayed diagnosis with conventional NBS tests for high-risk neonates without pre-selection of specific phenotypes in the NICU.
MATERIALS AND METHODS
Ethical considerations
The study was approved by the Institutional Review Board of Yonsei University Health System (IRB, 42017-0127). Korean neonates or infants who were admitted to the NICU at Severance Children's Hospital (Seoul, Korea) from May 2017 to September 2017 were included in the study after obtaining written informed consent from at least one biological parent/guardian. None of the babies was selected for metabolic disorders in line with the population-based NBS program.
Sample preparation, sequencing, and analysis of the TNGS panel and NBS
A TNGS panel for neonatal diseases was designed and validated in a previous study.12 A targeted gene enrichment method was used to construct libraries for subsequent determination of sequences using an NGS method with HiSeq2000 (Illumina, San Diego, CA, USA). In this study, the probe set was designed to capture 1.45 Mb, covering the exons and 25 nucleotides at the flanking introns for 198 targeted genes (Supplementary Table 1, only online). Probe-library hybridization followed by capture of target genes was performed according to the manufacturer's instructions.
A total of 48 samples were registered for the TNGS panel, which when run together using the developed panel of a 1-Mb region with a 1-Gb output would likely cover ≤×500 reads. We used blood samples collected on filter paper, and DNA was isolated from dried blood spots with QIAamp DNA Mini Kits (Qiagen, Valencia, CA, USA). The DNA was enzyme-fragmented with NEBNext dsDNA Fragmentase® (New England Biolabs, Ipswich, MA, USA), which was used to construct sequence libraries, according to the protocol provided by Cellemix (Seoul, Korea). For library enrichment, we used a custom probe set synthesized by Cellemix (Seoul). The subsequent capturing procedure was performed with the MYbaits® kit (Arbor Biosciences, Ann Arbor, MI, USA), and sequencing was performed on a HiSeq 2000 system (Illumina).
BaseSpace (Illumina) was used for data processing, alignment, variant calling, and annotations. The average coverage of target bases was 99.8%, with 98.1% of the regions of interest having at least 10X coverage and 95.4% having at least 20% coverage (Supplementary Table 2, only online). The sequenced reads were mapped to the human reference genome (UCSC hg19) with Burrows-Wheeler Aligner (BWA-0.7.7-isis-1.0.0), and variants were identified with the Genome Analysis toolkit (GATK version 1.6.23-gf0210b3). After filtering out variants using an internal database, variants with a minor allele frequency of less than 1% in either the 1000 Genomes Project or Exome Aggregation Consortium database were assessed further. The criteria for classification of variants were based on the principle recommended by the American College of Medical Genetics and Genomics (ACMG) standards.13
In Korea, NBS is mandated by law, and initial NBS samples were collected based on conventional processes within 7 days with TNGS samples consecutively. For newborns weighing less than 2000 g or those starting TPN, repeated screening tests for 28 days after birth or before discharge were required. Abnormal NBS was conducted once more and considered presumptively positive. Additional biochemical tests were then performed based on ACMG ACT sheets (https://www.acmg.net), and medical records were reviewed.
RESULTS
Participant demographics
To evaluate the usefulness of the TNGS panel in NBS for neonates showing high numbers of FPRs, we tested 48 clinical samples for newborn diseases associated with 198 genes (Supplementary Table 1, only online). The individual participants in the NICU were not selected for metabolic diseases in advance and included both full-term and preterm infants. In this study, there were 18 (38%) preterm infants and 27 (56%) newborns with a birth weight of less than 2000 g (Table 1). The average birth weight was 1998 g (range: 610–3790 g).
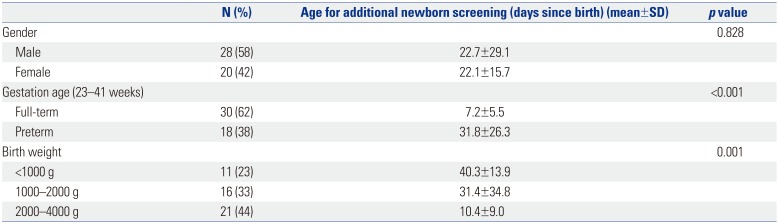
Table 1
Demographic Characteristics of Patients (n=48)
![]()
In our hospital, additional NBS tests are routinely performed to obtain more reliable results when babies have had 48 h of oral feeding after TPN is discontinued. Among the infants in the NICU that underwent additional NBS, those born with low birth weight (<2000 g) and preterm babies were assessed at later ages (days since birth) than full-term neonates to minimize the FPRs (Table 1).
Variants in the TNGS panel
The clinical characteristics of all newborns included in this pilot study are summarized in Supplementary Table 3 (only online). A total of 25 variants were identified in 19 patients (Table 2). In accordance with ACMG guidelines, 13 variants classified as “likely pathogenic” or “pathogenic” were detected in 11 (22.9%) of the neonates. In Korea, an extended NBS test using MS/MS was performed to detect approximately 54 metabolic diseases in a conventional NBS program. However, we identified pathogenic or likely pathogenic variants reported in six genes, including those associated with metabolic diseases not covered in the NBS: DHCR7 (Smith-Lemli-Opitz syndrome), PCBD1 (BH4-deficient hyperphenylalaninemia), GAA (glycogen storage disease II), ALDOB (fructose intolerance, hereditary), ATP7B (Wilson disease), and GBA (Gaucher disease). Two pathogenic GAA variants were identified in patient D17; however, those alleles were found in the same strand in the raw data and were negative in the enzyme analysis, suggesting a diagnosis as a carrier of Pompe disease. Of the known pathogenic mutations of the CYP21A2 gene, p. Gln319Ter is one of the most common variants related to congenital adrenal hyperplasia in Koreans, and the heterozygote mutation was identified in two unrelated patients in this study.14 One of the patients, D30, a female with ambiguous genitalia, showed normal 17-OHP levels in the NBS screening test, and the electrolyte balance was also normal. The other newborn, D43, was a male who underwent a colostomy operation due to a congenital imperforate anus.
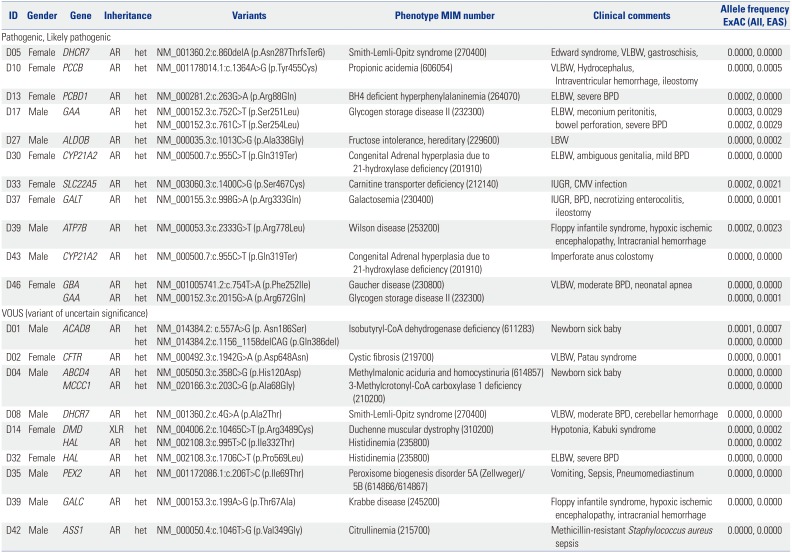
Table 2
Details of Variants Identified
MIM, Mendelian Inheritance in Man; ExAC, exome aggregation consortium; EAS, East Asian; AR, autosomal recessive; XLR, X-linked recessive; VLBW, very low birth weight (<1500 g); ELBW, extremely low birth weight (<1000 g); LBW, low birth weight (<2500 g); IUGR, intrauterine growth retardation; BPD, Bronchopulmonary dysplasia; CMV, cytomegalovirus.
![]()
Three of the newborns (D01, D03, D04) showed positive results in a comprehensive NBS. In the case of D01, elevation of C4 using MS/MS resulted in suspicion of an isobutyl-CoA dehydrogenase (IBDH) deficiency and short-chain acyl-CoA dehydrogenase deficiency. According to the ACMG algorithm, ethylmalonic encephalopathy (associated with the ETHE1 gene) is one of the diseases included in the differential diagnosis NBS.15 As a result, two mutations in ACAD8 were identified in patient D01, and the patient was ultimately diagnosed with IBDH deficiency associated with a novel compound heterozygous variant. For patients D03 and D04, increases in C5-OH acylcarnitine (3-OH isovalerylcarnitine) were reported to result in the same suspected disease of 3-methylcrotonyl-CoA carboxylase deficiency inherited in an autosomal recessive pattern. However, in the former patient, a heterozygote mutation (NM_020166.3: c.1391A>C; p. His464Pro) was identified in the MCCC1 gene, which is reported to be benign. In the latter case, D04, one unknown variant of the MCCC1 gene was identified and classified as a variant of unknown significance. In addition, biotinidase deficiency (associated with the BTD gene), which is one of the diseases causing elevated C5-OH, is not included in the Korean screening program at present, but could be evaluated through the TNGS panel. However, in patient D04, considering the limitations of not finding additional variants, such as deletion or duplication, it is necessary to confirm the clinical characteristics of the patient, and additional biochemical tests may be needed. Ultimately, the abnormal NBS tests were considered presumptively false-positive in these two asymptomatic patients based on assessment with the TNGS panel.
DISCUSSION
In this pilot study, we used a TNGS panel as a supplementary method for the conventional NBS program to achieve early diagnosis and to determine cases of false positive and carrier status in newborns in the NICU. This expands the applications of a few previous studies in which genetic confirmation tests were used only for neonates with abnormal NBS results or symptomatic disorders.371617
The NBS program is a public health program in Korea aimed at screening every neonate for inherited metabolic disorders and was introduced to identify conditions that can be critical to a child's health and survival. However, the screening program often produces high number of FPRs, which means that a baby with a suspected diagnosis may not always have health problems. To correctly identify all affected neonates, a confirmatory test, including biochemical analyses, enzyme activity, and genotyping, is required for positive results determined with NBS. In addition, FPRs (about 5–20%) are more common for infants in the NICU, or those with low birth weight, that are ill, and on TPN, which require additional follow-up NBS and confirmatory testing. The FPRs were reported to be 13-fold higher in very low birth weight infants (1000 to 1499 grams), and at least one in 10 of those infants showed FPRs with NBS. If the blood collection is delayed until the infant is completely off TPN, it could result in delayed diagnosis for an infant born with a low birth weight and/or preterm baby in NICU.345
To resolve these issues, we introduced TNGS to screen stressed newborns in the NICU undergoing the constitutional NBS program (Supplementary Fig. 1, only online).5615 Dried blood spot samples initially collected from these infants at high risk of FPRs were sent for NBS and the TNGS panel simultaneously. NBS was conducted based on the ACMG ACT sheet and algorithm as the conventional test, followed by interpretation of genetic variants according to ACMG standards. When pathogenic, likely pathogenic, or variants of unknown significance were identified in the infants, the concordance between other confirmatory tests, such as biochemical tests, and variant classification was evaluated.
This is the first application of the TNGS panel in conjunction with conventional population-based NBS for infants in the NICU to reduce the rate of FPRs and diagnose the precise metabolic disorder. This application might have broad implications for changing practice in the NICU or cardiac intensive care unit to enable a faster diagnosis and thus allow for more timely intervention. In addition, the repetitive testing without an accurate diagnosis of suspected disease imposes a huge amount of stress on the family because metabolic disorders can be fatal in some cases. Moreover, individuals with one copy of a recessive allele could be identified with the TNGS panel, allowing for identification of carriers with an asymptomatic condition that might warrant regular follow-up.
A definitive diagnosis was made in only one (D01) of the 48 infants included in this pilot study, which reflects the overall rarity of metabolic disorders. IBHD deficiency is an inborn error of valine metabolism with increased C(4)-carnitine levels, as identified by MS/MS. Most cases of IBHD deficiency were asymptomatic, and very few affected patients presented anemia, failure to thrive, dilated cardiomyopathy, and plasma carnitine deficiency. The patient D01 showed no related symptoms, and regular check-ups were performed. Because the range of symptoms of IBHD deficiency remains unclear, an exact diagnosis requires both NBS and molecular testing, such as TNGS. Two asymptomatic infants (D03 and D04) yielded FPRs, and the suspected diagnosis from the NBS was ruled out based on the mutations detected. However, patients with rare metabolic disorders sometimes have highly variable phenotypes, and hence, patients with abnormal NBS results should follow up their clinical symptoms and consider further evaluation if necessary.
Overall, FPRs and diagnostic yields of NBS cannot be estimated with this limited sample size. Moreover, the diagnostic yield seems relatively low because the actionable child-onset-genes related to NBS was designed for diagnosis of a rare genetic disorder with low incidence before clinical symptoms occurred. Also, newborns enrolled in this study were not limited to patients suspected of specific metabolic diseases, which is the first valuable pilot study showing that TNGS can be used to address the disadvantage of NBS screening. If the patients show clinical symptoms for suspected metabolic or genetic disorders while waiting for results of the TNGS test, the early diagnostic yield could be increased through a trio-based NGS test involving family history or parental samples.
In Korea, although the government-sponsored NBS program using MS/MS can screen over 50 diseases, some critical inherited disorders, such as glycogen storage disease, lysosomal storage disorder, severe combined immune deficiency disorder, adrenoleukodystrophy, and ornithine deficiency are not included. Therefore, rare metabolic disorders and several genetic disorders affecting infants and children could be screened before the onset of clinical signs using the TNGS panel.
Integration of our TNGS panel with NBS might help to avoid incidental findings and later-onset diseases rather than conducting whole-genomic sequencing at birth as part of the NBS program, and the specific genes included in the panel could be designed considering regional incidences based on ethnic background.1819 However, there is still room for improvement in the current TGNS panel, which does not cover deletion/duplication variants and deep intronic or promoter variations sufficiently, potentially resulting in increased false negative results. We also found that compound heterozygote conditions cannot be detected by the TNGS panel alone, owing to the technical limitation of distinction between cis or trans variants, except for mutations that are located very close to each other. as in infant D17.
Despite these limitations, we demonstrated that screening of ill infants with a TNGS panel could reduce delayed diagnoses, even for those at high risk of FPRs, using the current national NBS program. Moreover, disorders not detected in the current NBS can be diagnosed with the TNGS panel. The proposed practical workflow for conducting the TNGS panel analysis concurrently with initial NBS in stressed babies is expected to substantially reduce the number of unnecessary repetitive NGS tests and allow for the faster detection of rare metabolic disorders in infants.
 XML Download
XML Download