

 PDF
PDF Citation
Citation Print
Print




INTRODUCTION
The objective of medical device vigilance (MDV) is to improve protection of health and safety of patients, users, and others by reducing the likelihood of reoccurrence of incidents related to medical devices elsewhere. This can be achieved by evaluating adverse event reports and dissemination of information that may reduce the likelihood of adverse events, prevent repetition of adverse events, or alleviate consequences of such repetition.1 Reporting of medical device adverse events may easily be made by healthcare workers who are using medical devices to perform care activities on patients. According to the experiences in practice of monitoring medical device safety information within a healthcare institution, however, many healthcare workers do not report adverse events due to individual differences in judgment criteria, concerns about the disadvantages of reporting, and complaints about lack of time to report, etc. The results of a study on the perception and behavior of medical device safety in a tertiary care hospital in Korea have reported that healthcare workers need education on medical device safety due to lack of in-depth knowledge of adverse events of medical devices.2
Medical device adverse events are unexpected events that occur during or after patient use of a medical device. The World Health Organization (WHO) has defined an adverse event as “a problem that can or does result in permanent impairment, injury or death to the patient or the user.”3 It means we need two requirements to determine a medical device adverse event. The first is that the event occurs related to medical device usage. The second is that we need to determine whether the use of the medical device could cause direct harm or potential harm. Healthcare providers are well aware that all medical devices have expected or unexpected risks of adverse events when they apply these medical devices to patients. However, the use of the medical device in modern medicine is an inevitable choice. Sometimes, medical device problems can only be discovered after extensive market experiences are available.3
The International Medical Device Regulators Forum (IMDRF) is an international organization established in 2011. It aims to accelerate international medical device regulatory harmonization and convergence. Its management committee consists of 10 countries, including Korea, Japan, USA, China, and the EU.4 Medical device regulations of IMDRF member countries must comply with guidelines of the IMDRF and Global Harmonization Task Forces (GHTF), the predecessor of IMDRF. Asia-Pacific Economic Cooperation (APEC) has also recommended member economies to follow IMDRF or GHTF guidelines. The GHTF/SG2/N54R8:2006 guideline provides which events have been reported and how to report such events to the regulatory authority.1 Adverse events that can be reported to regulatory authorities in the use of devices specified in the GHTF/SG2/N54R8:2006 must meet the following three conditions. The first is that the event occurred with use of the medical device. The second is the association between the use of the medical device and the adverse event. The third is that the event has resulted in death or serious injury or the event might lead to death or serious injury if it recurs.1
Ideally, the application and interpretation of the international standards should not have differences by country or by the individual. In general, however, international standards and guidelines are presented as declarative norms; it means there needs to be a process of adaptation and implementation at the level of the individual country that is consistent with the intent of the standards under the national context. The Regulatory Harmonization Steering Committee (RHSC) of APEC has started the “Roadmap to Promote Regulatory Convergence for Medical Device Vigilance” since 2016.5 Its purpose is to reduce the possibility of recurrence of similar accidents by sharing information related to medical device's adverse events and contribute to the development of medical devices. As part of this effort, the National Institutes of Medical Device Safety Information (NIDS) in Korea in 2018 conducted an APEC MDV Center of Excellence (CoE) Pilot Training for regulatory authorities from APEC economies.6
The objective of this study was to report observational evidence that there were differences in recognizing and determining medical device adverse events. Such difference could contribute to underreporting of adverse events in real healthcare fields. We also identified the need for education and training to achieve consensus on regulations or guidelines.
METHODS
Introduction of MDV training courses for regulatory authorities in APEC economies
The NIDS was designated as a pilot CoE by APEC in 2017 about MDVs for APEC member countries. It conducted the first education and training program for regulatory authorities in cooperation with APEC Harmonization Center (AHC) in September 2018. Twenty-three regulatory authorities from 12 APEC economies (Chile, Mexico, Peru, Papua New Guinea, Indonesia, Thailand, Malaysia, Vietnam, Singapore, the Philippines, Hong Kong, and Chinese-Taipei) related to MDV participated in this training program. The demographic characteristics of the participating regulators are as follows. 17 (73.9%) were women and 6 were men (26.1%). Six (26%) were in their 50s, 5 (21.7%) were in their 40s, 7 (30.4%) were in their 30s, 2 (8.7%) were in their 20s, and 3 (13%) provided no response. The training program was designed with the aim to encourage harmonization of MDV regulations of APEC economies by training and applying MDV guidelines of the IMDRF and the GHTF.17 The curriculum consisted of education on contents of guidelines for medical device related adverse events, applying them individually to virtual cases, and making decisions in consensus through small-group discussions.5
Development of an educational tool for guideline application training
Question about the recognition of a phase of using a medical device for patients (patient use)
A medical device adverse event is defined as an unexpected event that occurs during or result from ‘patient use’ of a medical device. During experience with medical device adverse events monitoring, there is confusion in defining the beginning point of ‘patient use’ in medical practices. A close-ended question on a three-phase process of using medical device in healthcare fields was presented to each participant. Participants were asked to choose criteria applicable to their country. The three phases of patient use were defined based on the point at which differences in interpersonal opinions were issued frequently in discussions of individual cases on medical device adverse events in the medical device safety monitoring center network in Korea. These three phases are as follows. Phase 1 ‘inspecting’ means that problems are discovered while observing the product after placing the delivered medical device at the location where the patient is about to use it. Phase 2 ‘preparing’ means that problems are found during the preparation to perform the doctor's order but the device is not directly applied to the patient yet. Phase 3 ‘applying’ means that problems are found while directly applying the medical devices to the patient.
Development of virtual cases of medical device related adverse events
Six types of medical device related adverse events that could be experienced relatively frequently in the clinical field were selected and a piece of brief information necessary for the discussion by participants was constructed. MDV experts from Korea, Japan, and Malaysia participated in the development of virtual cases. Virtual cases covered the following:
Case 1. Infusion pump injection error: An analgesics mixture was set to be injected with 2 mL/h speed by infusion pump. The patient was found in an unconscious state by a nurse after 13 hours of infusion of the mixture. The nurse found the unconscious patient after 13 hours of injection. The IV bag was empty, but the settings of the infusion pump were the same as when it was started. The patient had supportive medical care like fluid therapy etc., and then recovered. Finally, the patient got back to his common daily life.
Case 2. Flowing foreign particles in IV fluid: The patient found flowing particles in the antibiotic mixed fluid. The pharmaceutical company collected three particles from the reclaimed materials and analyzed their physical and chemical characteristics. Two of those originated from the medication port's rubber packing of the IV fluid bag, and another one originated from the antibiotic vial's rubber packing.
Case 3. Problems in contact lens user: The patient was diagnosed as having a corneal ulcer by their contact lens.
Case 4. Removal of device fragments: A patient with varicose veins (men, 61-year-old) underwent surgical operation for removing the Saphenous vein by using a vein stripper under spinal anesthesia. The thoracic surgeon heard some clicking sounds from the operating spot, and then the surgeon recognized the silicon head broken apart from the vein stripper. The thoracic surgeon successfully found the stripper head using fluoroscopic X-ray imaging and removed it. The operation time was about 30 minutes longer than expected. Finally, the patient recovered and got back to his common daily life.
Case 5. Remained fragment of a device: A patient with varicose veins (men, 61-year-old) underwent surgical operation for removing the Saphenous vein by using a vein stripper under spinal anesthesia. The thoracic surgeon heard some clicking sounds from the operating spot, and then the surgeon recognized the silicon head broken apart from the vein stripper. In operating room, the medical team spent one hour trying to find the stripper head using fluoroscopic X-ray imaging, but they could not locate it. They completed the surgery then examined the patient’s body by using whole body computed tomography angiography scan. The broken piece was found in the inguinal area and removed by additional surgical operation with local anesthesia. Finally, the patient recovered and got back to his common daily life.
Case 6. Dislodged coronary stent: The surgeon felt slight resistance while pushing and pulling back on the stent in positioning. The stent dislodged from the balloon due to the resistance and was not visible. The stent was removed successfully without any patient injury. The procedure took 12 minutes longer than expected. The patient recovered and got back to his common daily life.
Development of questions for training GHTF or IMDRF guidelines
The purpose of the training was to achieve regulatory harmonization of the IMDRF or GHTF guideline for medical device adverse events among APEC economies. The intention was to reflect specific contents of the IMDRF/National Competent Authority Report (NCAR) WG/N14FINAL:2017 (Edition 2) and GHTF/SG2/N54R8:2006. We developed a total of six close-ended questions, including whether a medical device adverse event occurred or not, medical device relevance, health impacts, use errors, and whether there was value to share adverse event information across countries (Table 1). The intention of making the closed-ended questions was to clearly show the differences in interpreting and applying international guidelines from the IMDRF/GHTF. We think this not only facilitates discussions to resolve differences between individuals but also makes it easy and clear to see differences in opinions between groups.
Table 1
Questions that were applied in virtual cases for training of medical device adverse events

NCA = National Competent Authority, NCAR = National Competent Authority Report, GHTF = Global Harmonization Task Force, SG = study group, IMDRF = International Medical Device Regulators Forum, WG = working group.
![]()
Allocation of participants and virtual cases for discussion and consensus building
Twenty-three regulatory authorities from 12 APEC economies participated in the training. Participating countries were as follows: Chile, Mexico, Peru, Papua New Guinea, Indonesia, Thailand, Malaysia, Vietnam, Singapore, the Philippines, Hong Kong, and Chinese-Taipei. In the course of the education program, MDV experts and regulatory authorities from Korea, Japan, and Malaysia served as facilitators of education and discussion. Participants were allocated into six discussion groups and two virtual cases were assigned per group. After giving participants time to answer questions about virtual cases individually, group discussion was conducted. Each group leader presented their agreed results after their group discussion. One virtual case was discussed independently in two groups. All participants referenced the same guidelines for each question. Table 2 presents raw data for the assigned virtual cases and agreed results of the six groups. We partially applied team-based learning format to our program.8 We distributed relevant GHTF and IMDRF guidelines to participants two weeks before the CoE training. On the CoE training day, an education session about relevant GHTF and IMDRF guidelines was provided before group discussion using virtual cases.
Table 2
Results of group discussion on questions related to medical device adverse events

NCA = National Competent Authority, NCAR = National Competent Authority Report, IV = intra-venous.
Bold/Italic font = It shows case study results that the opinions of the two groups are inconsistent.
![]()
RESULTS
Recognition of the beginning point of ‘patient use’
We received responses from a total of 14 countries, including 12 CoE participating countries plus an additional two economies (Korea and Japan) who were facilitators of the education program. As for the question of “when is a beginning point to define ‘patient use’ of medical devices,” four (28.6%) countries answered phase 1, five (35.7%) countries answered phase 2, and five (35.7%) countries answered phase 3 (Fig. 1). This suggests that there is no consensus among APEC economies about the beginning point recognition of ‘patient use.’
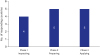 | Fig. 1Results of the survey for recognition of the beginning point of ‘patient use’. Phase 1 ‘Inspecting’: Problems discovered while observing the product after placing the delivered medical device at the location where the patient is about to use it; Phase 2 ‘Preparing’: Problems found during the preparation to perform the doctor's order without directly applying the medical device to the patient; and Phase 3 ‘Applying’: Problems found during direct application of medical device to the patient.
|
Results of group discussion on questions related to medical device adverse events
Only for question 1, there was perfect agreement between the two discussion groups for all six virtual cases. This meant that all developed cases were appropriately selected and developed to discuss medical device adverse events. For question 2, a question about the association of medical devices with adverse event, a different opinion for case 2 was observed between discussion groups. For question 3, a question about the health impact of the event, different opinions for virtual cases 1 and 4 were found between discussion groups. For question 4, a question about exemption rules for National Competent Authority (NCA) reporting, different opinions for virtual cases 2 and 3 were found between discussion groups. For question 5, a question about reporting to the NCA of adverse events related to use error, different opinions for virtual cases 2 and 4 were observed between discussion groups. For question 6, a question about the value of the exchange of NCAR to sharing adverse event information across countries, only for case 1 was the same opinion observed between discussion groups. These observed results confirmed that all participating groups made different judgments for the same event except for the question about a reportable medical device adverse event, although they applied the same criteria written in the guidelines (Fig. 2).

DISCUSSION
No matter how robustly medical devices are manufactured, they cannot guarantee zero risk of failure. Adverse event monitoring of medical devices at healthcare practices is crucial for securing health safety of patients and improving medical devices so that they could become useful for the development of new devices. Related surveillance activities such as the MedSun in USA, the Canadian Medical Devices Sentinel Network (CMDSNet) program in Canada and the IRIS inSite project in Australia have demonstrated that many countries are aware of the importance of monitoring adverse events associated with medical devices and participation of healthcare professionals as effective means to decrease the risk of harm caused by medical devices and strengthening public health.91011 In Korea, with the support of the Ministry of Food and Drug Safety (MFDS) in 2011, adverse event monitoring of medical institutions was started at six general hospitals that have a role as regional centers across the country. In 2019, it was expanded to 19 general hospitals that have a role as regional centers across the country. These regional centers are building a cooperative system with 740 healthcare institutions as a satellite monitoring site in the surrounding area. Adverse events reporting from medical institutions have increased from 128 in 2011 to 5,080 in 2018, although its benefit has not yet been fully reflected in the medical field.
The rate of official reporting of medical device malpractice cases was less than 0.5% according to the report of the Food and Drug Administration (FDA).12 The Australian Therapeutic Goods Administration (TGA) reported that adverse event rates associated with the use of ventilator and urogynecological mesh were less than 0.4% and 1.5%, respectively.13 Underreporting of medical device adverse events means loss of opportunities that can be used as information to prevent reoccurrence of the same events or possibilities that can be handled as relevant information for the development of medical devices.14 TGA has reported that the unreporting culture and the lack of awareness of adverse events in healthcare professionals and related stakeholders are the major causes of underreporting.13 Polisena et al.15 have performed a systematic review and suggested the following four main barriers in a hospital setting to event reporting: fear of punishment or censure, uncertainty regarding what should be reported, how incident reports will be used, and lack of time.
We observed that there was a considerable difference in judgment regarding the beginning point of patient use of medical devices across countries. Such difference is a critical issue in terms of international harmonization of MDVs. Although the same event is observed at the same time point, it is judged as an adverse event in some countries, but not in other countries. This might have happened because stakeholders of medical devices have no chance to discuss this topic.
Through a training course using virtual cases, we found that there was also a significant difference in applying definitions of adverse events written in guidelines of medical devices. The “uncertainty regarding what should be reported” is due to the lack of knowledge of medical device adverse events that can lead to underreporting. However, our observation implies that there is a “lack of consensus” due to perspective diversity in the actual application of guidelines even though participants might have the same knowledge given by sufficient provision of education.
For question 2 about the application of exemption rules, we observed differences between two discussion groups for virtual case 2. The intention for discussion in the development of virtual case 2 was to discuss the process to find out whether there was a certain area of pharmacovigilance (PV) or MDV by country. This issue should also be recognized as an adverse event beyond the problem of PV or MDV. Our expectation is that there will be differences in judgment by country. It will be the subject of discussion in the dimension of MDV harmonization. However, these two groups that discussed this virtual case included participants from the same country. Thus, individual opinion rather than country-specific principles might have influenced group judgment.
Three of the six virtual cases we developed were about detections and resolutions of incidents by health professionals at the site the events occurred. In virtual case 5, additional invasive interventions were required for resolution of the incident. In question 3 about health impact of the event for virtual case 5, both discussion groups answered serious injury. In virtual cases 4 and 6, on the other hand, remedial actions were relatively quick and immediate after the occurrence of the event. In the development of two virtual cases, the intention we proposed to participants as a topic of discussion was about the time spent in remedial action. For virtual case 6, which took an additional 12 minutes for the remedial action, the two groups answered as a near incident. However, for virtual case 4 that consumed 30 minutes more, the two groups presented their decision differently (as serious injury or near incident). In virtual case 1, two groups presented different answers (as near incident or serious injury). The group that answered ‘near incident’ explained the reason as follows: “In conclusion, the patient did not die or have a serious injury.” and “There was no further damage to the patient. The problem was nearly solved.” Their focus on judgment of health impact might be based on healthcare the professional's remedial action and patient's final condition. However, participants in favor of ‘serious injury’ mentioned the following reasons: “The patient was not dead, but was in a coma for a while.” and “If this happens again, it can lead to serious injury or death to the patient.” It seemed that they focused on medical device adverse event occurrence progress and potential risk rather than results of the medical device adverse event. These results suggest that the potential harms when it recurs and the invasiveness of remedial action can affect the judgment of an adverse event, even if the incident does not affect the patient's final health outcome.
The six groups that judged health impact of the event as near incident were equally divided on the application of exemption rule. On the other hand, all groups but one presented their opinion as disagreeing with applying the exemption rule if the group judged the health impact of the event as a serious injury. It seems that participants decide to apply the exemption rule in consideration of adverse event when the severity of health impact is considered the most. They also considered other conditions such as potential risk and expected adverse event. The intention for virtual case 3 was to obtain judgment when the incident that occurred was associated with the use of the medical device by the patient himself/herself and failure to comply with doctor's instructions. According to the GHTF guideline, the exemption rule is applied when it corresponds to the expected and foreseeable side effects. Nonetheless, the patient in this case developed a corneal ulcer and participants seemed to have taken the resulting health outcome as an important judgment.
In question 5, if the adverse event occurred by ‘use error,’ 8 of 12 responses were changed from all reportable events to non-reportable. Therefore, it is also necessary to clear the definition of ‘use error.’ Question 6 shows an apparent discrepancy about whether the adverse event could be included in NCAR exchange program.
The possible root cause of disagreement on specific questions can be attributable to many factors, such as the following: 1) limitation of declarative norms of the international guideline itself, 2) possible misunderstanding owing to language barrier, 3) difference of medical device-related regulatory system in participants' countries, and 4) perceptional differences. Except for discussion of differences in language barriers or medical device-related regulatory systems in individual countries, we identified through our observed results that we need to overcome differences in recognition and application of recommendations to international standards and create consensus.
Existing studies related to the reporting of medical device adverse events in MDV have been conducted mainly in terms of medical device users' awareness, reporting systems, and organizational culture that allow reporting.21617181920 However, there are no actual observational reports that the perspective diversity can affect the judgment of an adverse event as shown in our result. Because these results were observed from the training process of MDV, we could not investigate factors affecting individual point of view such as participants' personal career or their national regulation. It is also difficult to generalize the results of this study because all participants are regulatory authorities and the number of subjects is small. The strength of this study is that 1) structured definitions were used for discussions about the beginning point of patient use and 2) it showed the existence of perceptional differences in applying the established regulations to actual reality, using virtual cases with close-ended questions.
In conclusion, we identified perspective differences in recognizing and determining medical device adverse events. The perspective difference is one possible factor that might contribute to the underreporting of adverse events in real healthcare fields. The way to overcome perspective differences will be continuous education and training for the formation of consensus on international standards and guidelines. It is crucial to educate healthcare workers because they are in the first-line to recognize the adverse events. Then, we believe the training method we developed can be useful and readily shared globally. It will not only increase the accuracy and reliability of information exchanged about medical device adverse events between national competent authorities, but also ensure patient safety and prevent the occurrence of medical device adverse events.
 XML Download
XML Download