

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Zika virus (ZIKV) is one of the pathogens which is transmitted world widely, but there are no effective drugs and vaccines. Whole genome sequencing (WGS) of viruses could be applied to viral pathogen characterization, diagnosis, molecular surveillance, and even finding novel pathogens. We established an improved method using direct RNA sequencing with Nanopore technology to obtain WGS of ZIKV, after adding poly (A) tails to viral RNA. This established method does not require specific primers, complimentary DNA (cDNA) synthesis, and polymerase chain reaction (PCR)-based enrichment, resulting in the reduction of biases as well as of the ability to find novel RNA viruses. Nanopore technology also allows to read long sequences. It makes WGS easier and faster with long-read assembly. In this study, we obtained WGS of two strains of ZIKV following the established protocol. The sequenced reads resulted in 99% and 100% genome coverage with 63.5X and 21,136X, for the ZIKV PRVABC59 and MR 766 strains, respectively. The sequence identities of the ZIKV PRVABC59 and MR 766 strains for each reference genomes were 98.76% and 99.72%, respectively. We also found that the maximum length of reads was 10,311 bp which is almost the whole genome size of ZIKV. These long-reads could make overall structure of whole genome easily, and WGS faster and easier. The protocol in this study could provide rapid and efficient WGS that could be applied to study the biology of RNA viruses including identification, characterization, and global surveillance.
Go to : 
REFERENCES
1). Malone RW, Homan J, Callahan MV, Glasspool-Malone J, Damodaran L, Schneider Ade B, et al. Zika Virus: Medical Countermeasure Development Challenges. PLoS Negl Trop Dis. 2016; 10:e0004530.

2). Sikka V, Chattu VK, Popli RK, Galwankar SC, Kelkar D, Sawicki SG, et al. The emergence of zika virus as a global health security threat: A review and a consensus statement of the INDUSEM Joint working Group (JWG). J Glob Infect Dis. 2016; 8:3–15.
3). Grard G, Caron M, Mombo IM, Nkoghe D, Mboui Ondo S, Jiolle D, et al. Zika virus in Gabon (Central Africa)–2007: a new threat from Aedes albopictus? PLoS Negl Trop Dis. 2014; 8:e2681.
4). Brasil P, Pereira JP Jr, Moreira ME, Ribeiro Nogueira RM, Damasceno L, Wakimoto M, et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N Engl J Med. 2016; 375:2321–34.
5). Bearcroft WG. Zika virus infection experimentally induced in a human volunteer. Trans R Soc Trop Med Hyg. 1956; 50:442–8.
6). Ahlfors K, Ivarsson SA, Harris S. Report on a long-term study of maternal and congenital cytomegalovirus infection in Sweden. Review of prospective studies available in the literature. Scand J Infect Dis. 1999; 31:443–57.
7). Schuler-Faccini L, Ribeiro EM, Feitosa IM, Horovitz DD, Cavalcanti DP, Pessoa A, et al. Possible Association Between Zika Virus Infection and Microcephaly – Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016; 65:59–62.
8). Mlakar J, Korva M, Tul N, Popović M, Poljš ak-Prijatelj M, Mraz J, et al. Zika Virus Associated with Microcephaly. N Engl J Med. 2016; 374:951–8.
9). Oehler E, Watrin L, Larre P, Leparc-Goffart I, Lastere S, Valour F, et al. Zika virus infection complicated by Guillain-Barre syndrome–case report, French Polynesia, December 2013. Euro Surveill. 2014; 19.
10). Broutet N, Krauer F, Riesen M, Khalakdina A, Almiron M, Aldighieri S, et al. Zika Virus as a Cause of Neurologic Disorders. N Engl J Med. 2016; 374:1506–9.
11). Houldcroft CJ, Beale MA, Breuer J. Clinical and biological insights from viral genome sequencing. Nat Rev Microbiol. 2017; 15:183–92.
12). Grad YH, Newman R, Zody M, Yang X, Murphy R, Qu J, et al. Within-host whole-genome deep sequencing and diversity analysis of human respiratory syncytial virus infection reveals dynamics of genomic diversity in the absence and presence of immune pressure. J Virol. 2014; 88:7286–93.
13). Parker J, Chen J. Application of next generation sequencing for the detection of human viral pathogens in clinical specimens. J Clin Virol. 2017; 86:20–6.
14). Baronti C, Piorkowski G, Leparc-Goffart I, de Lamballerie X, Dubot-Pé rè s A. Rapid next-generation sequencing of dengue, EV-A71 and RSV-A viruses. J Virol Methods. 2015; 226:7–14.
15). Marston DA, McElhinney LM, Ellis RJ, Horton DL, Wise EL, Leech SL, et al. Next generation sequencing of viral RNA genomes. BMC Genomics. 2013; 14:444.
16). Oude Munnink BB, Kik M, de Bruijn ND, Kohl R, van der Linden A, Reusken C, et al. Towards high quality real-time whole genome sequencing during outbreaks using Usutu virus as example. Infect Genet Evol. 2019; 73:49–54.
17). Bowden R, Davies RW, Heger A, Pagnamenta AT, de Cesare M, Oikkonen LE, et al. Sequencing of human genomes with nanopore technology. Nat Commun. 2019; 10:1869.
18). McNaughton AL, Roberts HE, Bonsall D, de Cesare M, Mokaya J, Lumley SF, et al. Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV). Sci Rep. 2019; 9:7081.
19). Wongsurawat T, Jenjaroenpun P, Taylor MK, Lee J, Tolardo AL, Parvathareddy J, et al. Rapid Sequencing of Multiple RNA Viruses in Their Native Form. Front Microbiol. 2019; 10:260.
20). Garber K. Epigenetics comes to RNA. Science. 2019; 365:16–7.
21). Lichinchi G, Zhao BS, Wu Y, Lu Z, Qin Y, He C, et al. Dynamics of Human and Viral RNA Methylation during Zika Virus Infection. Cell Host Microbe. 2016; 20:666–73.
Go to :
 | Figure 1.Sequence alignment display on the reference ZIKV genome with Geneious Prime for three runs. (A) Sequence alignment for ZIKV PRVABC59 strain from the first run. (B) Sequence alignment for ZIKV PRVABC59 strain from the second run.(C) Sequence alignment for ZIKV MR 766 strain from the third run. |
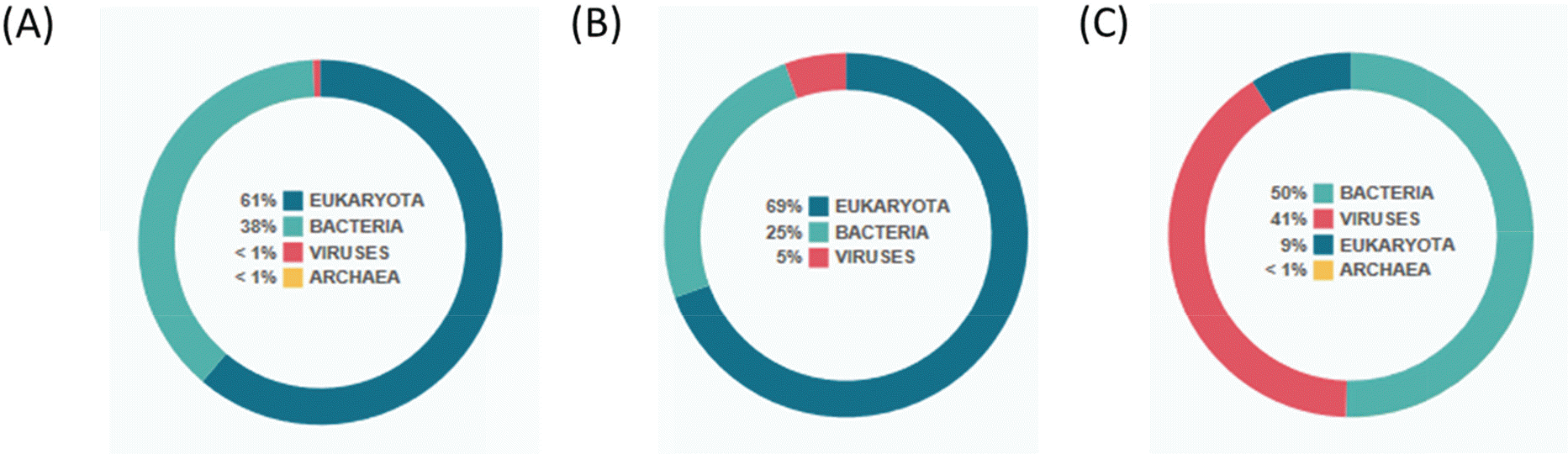 | Figure 2.Analysis with EPI2ME. EPI2ME is a cloud-based data analysis platform of nanopore data in real-time. It shows the portion of reads for each microorganism. (A) The portion of reads for ZIKV PRVABC59 strain from the first run. (B) The portion of reads for ZIKV PRVABC59 strain from the second run. (C) The portion of reads for ZIKV MR 766 strain from the third run. |
 | Figure 3.Long-reads with Nanopore technology. Several long sequences of ZIKV MR 766 were aligned to the reference genome. The maximum long sequence was 10,311bp. |
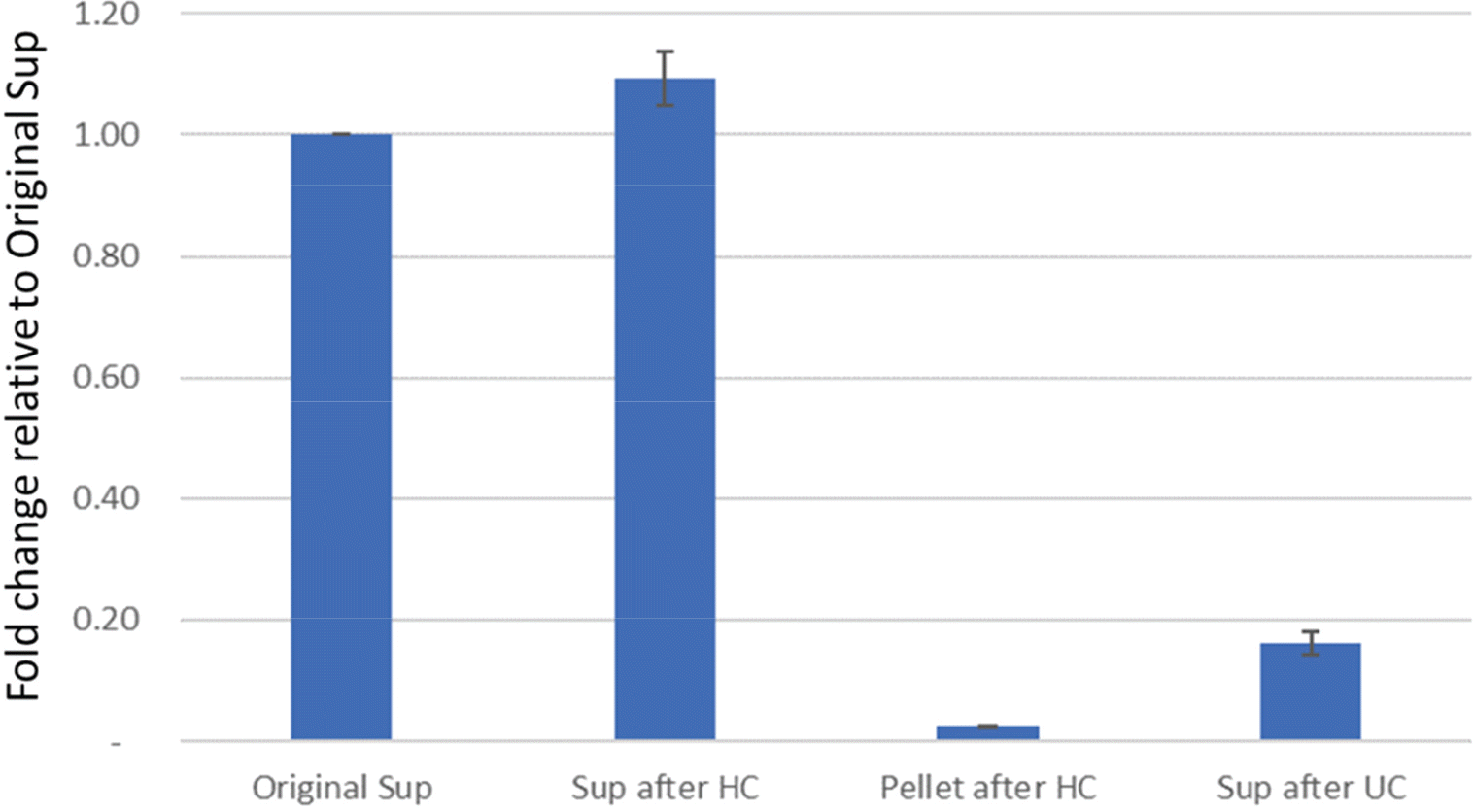 | Figure 4.Relative quantification of ZIKV. Original Sup represents the original viral supernatant, Sup after HC represents the viral supernatant after high-speed centrifugation, Pellet after HC represents the pellet after high-speed centrifugation of the viral supernatant, and Sup after UC represents the viral supernatant after ultra-centrifugation. Bars indicate SEM from three experiments. |
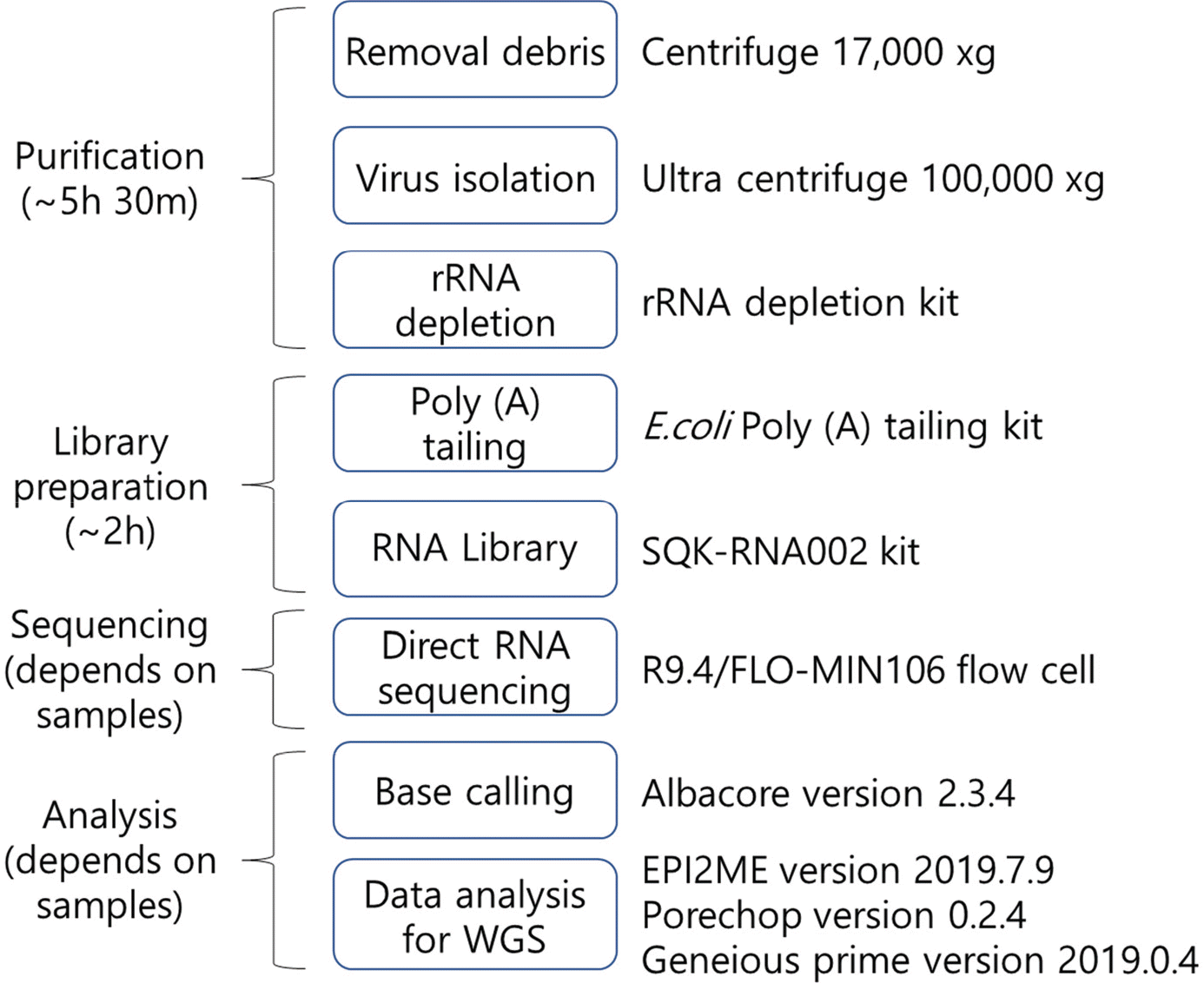 | Figure 5.The work flow for WGS of single-strand RNA virus using direct RNA sequencing with Nanopore technology. High-speed centrifugation was important to improve the efficiency of WGS and direct RNA sequencing with Nanopore technology could make WGS faster and easier with long-reads and without specific primers and PCR-based enrichment |
Table 1.
Sequence data after alignment
 XML Download
XML Download