

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Connective tissue disease (CTD) is a disorder characterized by diverse symptoms and signs as autoantibodies circulate in the system, causing damage to internal organs. Interstitial lung disease (ILD) associated with CTD is called CTD-ILD.
At an early stage in diagnosing ILD, it is very important to examine the patient with respect to the existence of various causes of ILD. Of the known causes of ILD, CTD is the most crucial. If ILD is diagnosed in a CTD patient and conversely, if CTD is diagnosed in an ILD patient, CTD-ILD is diagnosed. It is very critical to determine the existence of CTD in ILD patients, because there are significant differences between CTD-ILD and idiopathic interstitial pneumonia (IIP) in terms of both treatment approach and prognosis. Some CTD-ILD patients present with ILD prior to the CTD diagnosis. Therefore, even in those not previously diagnosed with CTD, any clinical symptoms suggestive of CTD, autoantibody test, and chest X-ray findings should be thoroughly checked to determine the existence of CTD.
Go to : 
Classification and Clinical Features of CTD-ILD
The incidence and clinical features of CTD-ILD differ depending on the form of the associated CTD. Across various types of ILD, the incidence of systemic sclerosis (SSc) is the highest, followed by idiopathic inflammatory myopathies (IIM), rheumatoid arthritis (RA), systemic lupus erythematous (SLE), and Sjögren's syndrome.
1. RA associated ILD
ILD is the most common among RA-associated lung diseases, but the prevalence varies depending on the patient group or diagnostic method1. A wide range of RA associated ILD (RA-ILD) prevalence rates, 19%–44%, has been reported in prospective studies using chest high resolution computed tomography (HRCT)2345, because each author used different ILD definitions and diagnostic methods. A cross-sectional study in which chest HRCT was performed at an RA clinic regardless of respiratory symptoms reported an ILD prevalence of 19%. In the study, chest HRCT had a higher sensitivity in diagnosing ILD than pulmonary function tests (PFT)6.
Although RA is more common among women, RA-ILD is reported to be more common in men. Some studies reported a male-to-female ratio of 2:178. Typically, it occurs in patients in their 40s and 50s, and age and smoking history are known risk factors of ILD91011. Rheumatoid factor and anti-cyclic citrullinated peptide were also found to be strongly associated with the occurrence of ILD12131415161718. In RA-ILD, the most common pathological finding in surgical lung biopsy is usual interstitial pneumonia (UIP)19.
2. SSc associated ILD
SSc, also called scleroderma, has a lower incidence but a higher level of pulmonary infiltration compared with RA. Pulmonary function abnormalities due to ILD are observed in as high as 70% of SSc patients, and about a third of the patients show clinically significant evidence of ILD1. Chest HRCT is essential for diagnosing SSc associated ILD (SSc-ILD), and the most common chest HRCT finding is non-specific interstitial pneumonia (NSIP)20. In general, ILD is common in patients with diffuse SSc, a condition in which internal organs as well as the skin are involved, and the autoantibody profile was found to correlate with pulmonary infiltration212223. The autoantibody most commonly found in patients with diffuse SSc is anti-topoisomerase I, also known as anti-scl-70. It has been reported that ILD occurred in 85% of anti-scl-70 positive patients and that the antibody concentration level was correlated with the severity and activity of ILD2425.
Fibrotic NSIP is the most common pattern in the pathological findings of SSc-ILD, and cellular NSIP is found in less than 10% of patients. Typical features of idiopathic pulmonary fibrosis, such as severe fibrosis and fibroblastic foci, are observed in less than 30% of cases with UIP262728.
SSc-ILD patients are at greatest risk for ILD progression in the first 3–4 years (45%–55%), and some patients show rapid pulmonary function decline during this time. Accordingly, PFT should be performed on a regular basis. Treatment should be considered in SSc-ILD patients under the following conditions: (1) if at the time of the initial diagnosis of ILD, ILD involvement exceeds 20% in chest HRCT, or forced vital capacity (FVC) is less than 70% of the predicted value in PFT, or (2) if diffusing capacity of the lungs for carbon monoxide (DLCO) is over 15% or FVC over 10% in a follow-up PFT629.
3. Sjögren's syndrome associated ILD
Studies have reported that pulmonary infiltration occurs in 9%–24% of all patients with Sjögren's syndrome, and that abnormal findings in PFT, bronchoalveolar lavage (BAL), and chest HRCT were observed in 75% of patients with asymptomatic Sjögren's syndrome32333435. Generally, pulmonary infiltration manifests in the end stage of Sjögren's syndrome and rarely occurs as the first manifestation36.
In Sjögren's syndrome, not only interstitial tissues in the lung but the entire respiratory system could be infiltrated, and so the patient could also present with small airway disease such as follicular bronchiolitis, aside from ILD. In whatever form, 10-year mortality increases by more than 4 times in Sjögren's syndrome patients with pulmonary infiltration compared to those without32.
4. Mixed connective tissue disease associated ILD
Mixed connective tissue disease (MCTD) was defined in 1972 for the first time, and there is still a controversy around this category. Generally, it refers to an autoimmune disease with positive anti-U1 ribonucleoprotein antibody. Pulmonary infiltration is quite common in MCTD, found in 47%–78% of patients3940. According to the literature, severe ILD occurred in approximately 19% of MCTD patients, and it was associated with anti-Ro-52 antibody but not with disease duration or smoking history4142.
5. IIM associated with ILD
IIM include polymyositis (PM), dermatomyositis (DM), and clinically amyopathic dermatomyositis, in each of which a varying degree of skin, muscle, joint, and lung infiltration is present. IIM associated with ILD (IIM-ILD) shows a variety of disease progression patterns from asymptomatic to rapidly progressive4344. The prevalence of ILD in IIM is also found to vary considerably, between 20%–78%, and comorbidity and mortality rates are reported to generally increase if ILD co-exists45464748. In IIM-ILD versus other CTD types, ILD more frequently occurs as the very first manifestation before it progresses to a systematic disease49505152.
Antisynthetase syndrome (AS) is a specific subset of IIM, with positive antisynthetase antibodies associated with protein synthesis. It is a syndrome characterized by the co-occurrence of ILD and the presence of some of the following manifestations: fever, joint pain, Raynaud's phenomenon, and mechanic's hands. Of various muscle-specific antibodies, anti-Jo-1 antibody is most commonly used in the diagnosis. As can be seen in the inclusion of ILD as an essential element in diagnosing AS, the importance of ILD in assessment of prognosis is also great. Like other types of IIM-ILD, AS-ILD too shows diverse disease progression patterns. It has been reported that approximately 50% of patients develop acute respiratory failure515354.
6. SLE associated with ILD
In SLE, pulmonary infiltration occurs in as high as 33%–50% of patients. But, in most patients, the entire respiratory system (thoracic cavity, lung parenchyma, airway, and pulmonary vessels) is infiltrated, and pulmonary infiltration of the pattern shown in ILD tends to be relatively infrequent (1%–15%) compared to other types of CTD435556. It has been reported that the longer the duration of SLE (over 10 years), the higher the risk of ILD57 and that SLE is strongly associated with ILD in the presence of Raynaud's phenomenon, positive response to anti-U1 RNP antibody, sclerodactyly, nailfold capillary loops, and old age5859.
Go to :
Diagnosis of CTD-ILD
1. Assessment of disease history and the presence of CTD
2) Patients never diagnosed with CTD
In some patients, ILD appears as the first manifestation among diverse clinical features of CTD. Such cases are most commonly observed in IIM (approximately 10%–30%), and additionally, the pattern is reported in some RA patients, and also in SSc patients though very rare6465.
If a patient without notable disease history rapidly develops dyspnea over several weeks to several months and eventually experiences respiratory failure while chest HRCT shows a ground glass opacity that is gradually progressing, the possibility of CTD-ILD should be considered. Particularly, if the level of muscle enzymes (e.g., creatinine phosphokinase, and aldolase) increases, IIM should be suspected as the underlying disease65. Even in cases with chronic ILD, CTD could be the underlying disease. Hence, whether or not the patient presents any symptom or other manifestation suggestive of CTD, this possibility should be considered (Table 1)66. For instance, Raynaud's phenomenon, dry eye and mouth, and muscle weakness tend to have low specificity, but the specificity of mechanic's hands (Figure 1A) or Gottron's papules (Figure 1B) is high. Thus, if mechanic's hands or Gottron's papules are present, DM is highly likely to be an underlying disease67. Such systemic symptoms or other manifestations that raise a suspicion of CTD often occur in women under age 5068697071.
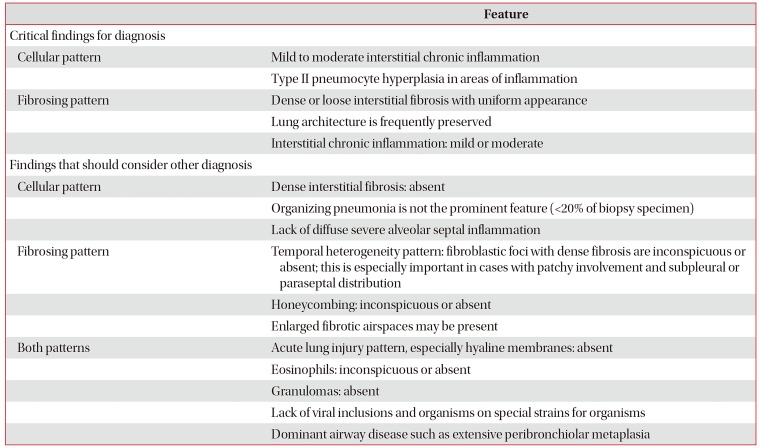
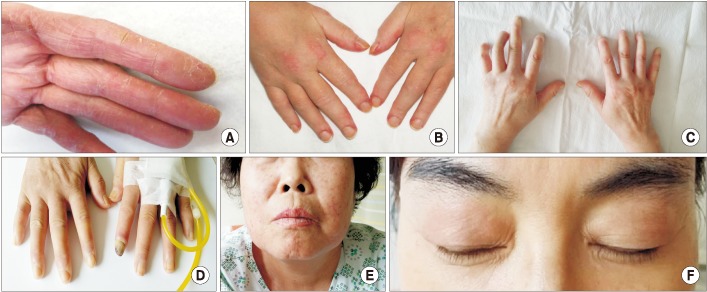 | Figure 1(A) Mechanic's hand, cracking and fissuring along the sides of the digits and palm. (B) Gottron's papules, red and scaly papules that erupt on the metacarpophalangeal joints. (C) Sclerodactyly, fixed fingers in a semi-flexed position with tightened and wax like skin. (D) Digital ulceration, an ulceration on the tip of index finger. (E) Telangiectasias, multiple dilated facial small vessels. (F) Heliotrope rash, violaceous erythema on the upper eyelids (The patient provided verbal consent for the picture).
|
Table 1
Core manifestations in ILD patients to diagnose underlying CTD66
| Organ | Manifestations to check |
|---|---|
| Peripheral circulation | Raynaud phenomenon |
| Skin | Sclerodactyly (Figure 1C) |
| Digital ulcerations or scars | |
| Telangiectasia (Figure 1D, E) | |
| Gottron's sign | |
| Heliotrope rash around the eyes | |
| Heliotrope rash in the neck, upper chest and shoulder area (Figure 1F) | |
| Photosensitivity | |
| Mechanics' hands | |
| Joint | Joint pain, joint swelling |
| Morning stiffness (over 60 minutes) | |
| Muscle | Muscle aches, muscle weakness |
| Mouth and eye | Dry mouth, dry eye (Sicca syndrome) |
![]()
2. Radiologic findings (chest HRCT findings)
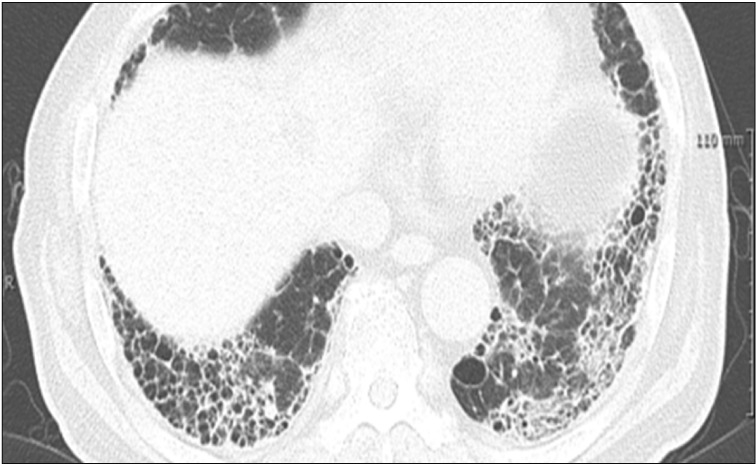
It is not possible to differentiate IIP and CTD-ILD based on radiologic findings. CTD-ILD often manifests itself in NSIP or organizing pneumonia (OP), or LIP (albeit rare) (Figure 2A–C), and IIP most often manifests itself in UIP (Figure 3). Thus, if a typical UIP is not found in an imaging test, CTD is likely to be the underlying disease60. An exception is that a radiologic finding of UIP is the most common in RA-ILD, and is often found in SSc-ILD, as well. The prognosis of RA-ILD is known to be poor in patients with a UIP pattern compared to those without UIP60.
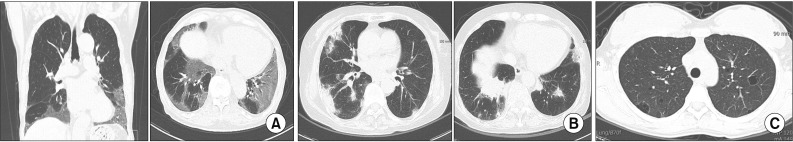 | Figure 2(A) Radiologic pattern of non-specific interstitial pneumonia in a patient with systemic sclerosis. High resolution computed tomography (HRCT) image shows bilateral subpleural and basal predominant fine reticular pattern and ground-glass opacity. (B) Radiologic pattern of organizing pneumonia in a patient with dermatomyositis. HRCT image shows multiple peripheral patch consolidations. (C) Radiologic pattern of lymphocytic interstitial pneumonia in a patient with Sjögren's syndrome. HRCT image shows multifocal variable-sized, thin-walled, cystic lesions on both lungs.
|
The radiologic pattern of ILD in CTD-ILD patients differs according to CTD type. In RA, UIP is the most common (50%–60%); in SSc, NSIP is the most common radiologic finding (80%–90%), although UIP is also found in as many as 10%–20%; in MCTD, NSIP is the most common; and, in PM and DM, NSIP, OP, UIP, and diffuse alveolar damage may be found65.
3. Surgical lung biopsy
It is still controversial whether surgical lung biopsy should be performed to confirm the diagnosis when CTD-ILD is suspected. The prognosis may differ depending on the pathological finding (UIP vs. others), and with biopsy, differential diagnoses of other diseases as well as ILD are possible. But, complications may occur following surgical lung biopsy, and thus the decision should be made by considering the pros and cons. In some patients, BAL is an alternative to biopsy, and using this procedure, infection can be excluded and the number of cells is estimated65.
Because a specific pathology does not exist in CTD-ILD, IIP and CTD-ILD cannot be completely differentiated based on pathological findings alone. However, because the proportion of NSIP is high in CTD-ILD, the presence or absence of underlying CTD should be looked for if NSIP is found on pathological analysis2872737475767778.
In addition, among those with CTD-ILD with a UIP pattern there were more germinal centers, lymphoid hyperplasia, plasma cells, and fewer fibroblastic foci and honeycombing, compared with cases of IIP with UIP. If the aforementioned features are found in conjunction with UIP, the presence or absence of CTD should be examined, because the co-occurrence of lymphocytic or follicular bronchiolitis is common (Figure 4)7980818283.
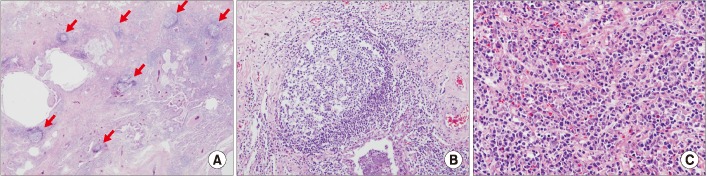 | Figure 4Histopathology of the lung in a patient with interstitial pneumonia with autoimmune features. (A) Usual interstitial with lymphoid follicles (×10). (B) Lymphoid bronchiolitis (×100). (C) lymphoplasmacytoid cell infiltrates (×200). Hematoxylin eosin saffron. Courtesy of Prof. Shim HS, Yonsei University.
|
4. Autoantibodies
When a patient is diagnosed with ILD, autoantibody testing should be performed to determine the likelihood of CTD-ILD. As shown in Table 2, cases exist in which an autoantibody is found but the clinical features do not correspond to the diagnosis of CTD or do not meet specific CTD diagnostic criteria. However, since such patients may present clinical features of CTD later, continuous follow-up is required and consultation with a rheumatologist for the symptoms is necessary6465.
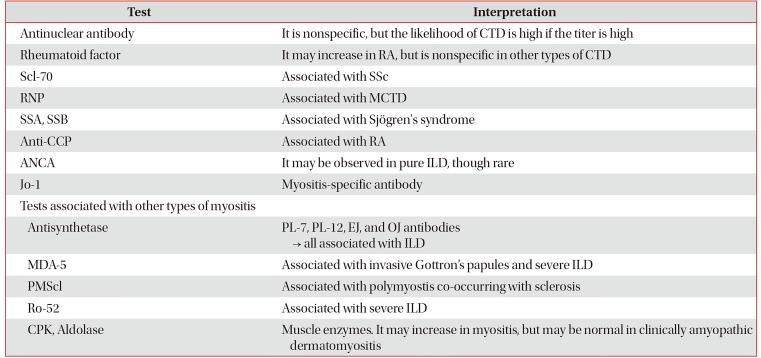
Table 2
Types and significance of autoantibodies65
![]()
Go to :
Treatment of CTD-ILD
1. Acute presentations of CTD-ILD
Acute presentation of CTD-ILD manifests itself in de novo acute interstitial pneumonia or an acute exacerbation of underlying CTD65. The treatment approach for acute exacerbation of CTD-ILD has not yet been established, but conventionally, it is treated using intravenous methylprednisolone (1 g/day intravenously for 3 days) followed by oral prednisolone (1 mg/kg/day). Cyclosporine (500–750 mg/m2 intravenously) and cyclophosphamide (CYC) (2–3 mg/kg/day) may additionally be administered (Table 3). Despite immunosuppressive therapy, patients presenting an acute exacerbation often need a ventilator and the mortality rate is high (83.3%)658485.

Table 3
Summary of acute presentation of CTD-ILD
![]()
2. Chronic presentations of ILD
The fundamental treatment of CTD-ILD is to tackle the primary cause of CTD. Evidence regarding treatment effect in CTD-ILD is lacking, because comparison group studies have not been conducted with the exception of SSc-ILD. Consensus has not yet formed regarding treatment initiation for CTD-ILD, but conventionally, a steroid or immunosuppressive drug is administered in patients in whom clinical features are severe, symptoms have recently started to progress, or the duration of systemic disease has been short86. Lung transplantation may be considered if the patient does not respond to drug therapy and ILD worsens.
1) Initial treatment of CTD-ILD
For the initial treatment of CTD-ILD, steroids are commonly used, but the recommendation as to which steroid is appropriate is undecided. The dose, route of administration, treatment duration, and dose tapering vary according to individual patients' clinical picture and the clinician's impression (Table 4)65. As the most common ILD form in IIM-ILD patients is OP, prednisolone 1 mg/kg/day is often used, i.e., the dose at which clinicians are relatively experienced in the treatment of cryptogenic organizing pneumonia (COP). However, treatment response to the therapy is reported to be low in the all forms of IIM-ILD except COP87.
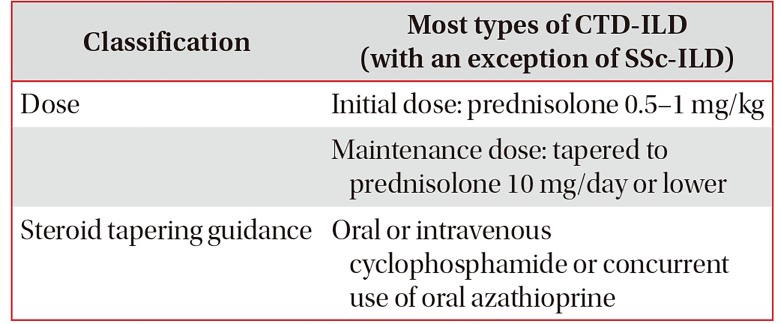
Table 4
Initial treatment of CTD-ILD85
![]()
Azathioprine (1–2 mg/kg/day) is often administered as combination therapy with a steroid to treat CTD-ILD, but evidence regarding monotherapy is insufficient88. A retrospective survival analysis conducted on 192 IIM-ILD patients showed lower mortality in the group administered with azathioprine (hazard ratio, 0.35; 95% confidence interval, 0.16–0.74; p=0.0064)47, but there is no evidence of an improvement in the survival rate in other forms of CTD-ILD.
CYC is the only immunosuppressive investigated in a randomized controlled trial. In SSc-ILD, FVC was improved after 1-year treatment in oral CYC (2 mg/kg/day) group compared with the control group89. Evidence on pulmonary function improvement is lacking in other types of CTD-ILD aside from SSc-ILD.
In a study with 125 patient cases with SSc-ILD, IIM-ILD, and RA-ILD, mycophenolate mofetil (MMF; 1.0–1.5 g bid) showed a reduction in the required dose of steroid and an improvement in FVC90. In addition, a recent randomized controlled trial with SSc-ILD patients demonstrated that the effect of MMF was similar to that of CYC91.
A retrospective study in IIM-ILD patients found that compared to the groups administered with CYC or cyclosporine in combination with a steroid, the group in whom tacrolimus was added had more favorable event-free survival92. The typical dose of tacrolimus is 1–3 mg/day65.
Currently, a randomized controlled trial with rapidly progressive CTD-ILD (including SSC-ILD, IIM-ILD, and MCTD-ILD) is under way to compare the effects of CYC and rituximab with FVC improvement as the primary endpoint93.
Table 5 summarizes treatment options for each CTD-ILD.

Table 5
Summary of treatment of CTD-ILD
![]()
2) Treatment of SSc-ILD
(1) PICO for treatment of patients with SSc-ILD: In SSc-ILD, is CYC more effective compared with placebo?
(2) Summary of recommendations for the treatment of patients with SSc-ILD: We suggest that the use of CYC may be considered in consultation with an expert, in some SSc-ILD patients experiencing dyspnea and reduced pulmonary function (weak recommendation, level of evidence; low).
FVC, total lung capacity (TLC), dyspnea, and quality of life index improved in the patient group administered with oral CYC over 1 year compared to placebo. A 1-year follow up after treatment termination showed that the improvement in dyspnea was maintained, but other indices returned to the levels prior to drug administration. Thus, evidence for long-term effectiveness of CYC is not sufficient, but it improved dyspnea and had a short-term effect in improving pulmonary function.
The effect of CYC in SSc-ILD patients has been reported in a considerable number of studies, but most of the studies were retrospective. Even the prospective studies tend to lack a comparison group or be observational. The authors conducted a systematic literature review on randomized placebocontrolled studies to investigate whether CYC has a positive effect on various SSc-ILD related indices such as the rate of pulmonary function decline, severity of dyspnea, quality of life, and functional ability. But the authors could not conduct meta-analysis because there was only one randomized placebo-controlled trial on CYC, i.e., Tashkin et al.'s Scleroderma Lung Study (SLS)89 published in N Engl J Med in 2006.
In SLS, 158 symptomatic SSc-ILD patients in whom disease activity was confirmed on either BAL or chest HRCT were randomly assigned to one of two groups, a group treated with CYC (2 mg/kg/day) for a year and a placebo group. During the study period, patients could be treated with prednisolone up to 10 mg/day. If an FVC reduction of 15% versus the baseline continued over 1 month from month 3 after treatment initiation, it was considered as treatment failure and the patient was unblinded and treated with CYC. The primary endpoint of the study was the FVC (expressed as a percentage of the predicted value) at 1 year after adjusting for baseline values and treatment group. Secondary endpoints were TLC, change in DLCO, disability index of health assessment questionnaire (HAQ), SF-36, transitional dyspnea index (TDI), and skin thickness score. Between-group difference in FVC change after 1 year of CYC administration was 2.53% (95% confidence interval, 0.28–4.79; p<0.03), indicating that treatment with CYC significantly slowed down the rate of FVC reduction. Of the secondary endpoints, TLC, HAQ, TDI, and skin thickness score were significantly improved in the CYC group in comparison to the placebo group.
Side effects of CYC that occurred during the study were hematuria (hemorrhagic cystitis), leukopenia, neutropenia, anemia, pneumonia, etc.8994. Therefore, it is recommended to regularly perform blood test and use mesna for the prevention of hemorrhagic cystitis, in patients treated with CYC. Long-term use of the drug is not recommended because of increased risk for bladder cancer and other malignant diseases.
After the completion of this SLS study, the researchers followed up the patients for an additional 1 year without administering a drug in order to investigate the duration of drug effect94. During the additional 1-year follow-up, if treatment failure (an FVC reduction over 15% compared to the baseline) occurred in either group, the patient was treated with CYC or an immunosuppressive according to the clinician's decision. During the 1-year follow-up, between-group differences in FVC and TLC were significant for the first 6 months, that is, the drug effect was maintained for a total of 18 months from the start of the study. But group difference disappeared by month 12 after treatment termination (that is, 24 months after the start of the study). An additional analysis showed a larger drug effect in patients with FVC less than 70%. On the other hand, the improvement of TDI (an index of dyspnea) was maintained up to month 12 after treatment termination94.
In summary, a year-long treatment with CYC administration improved pulmonary function, dyspnea, and quality of life and the effects were maintained up to 6 months after treatment termination. One year after treatment termination, two groups still showed a significant difference in dyspnea but not in FVC or TLC. Based on the improvement of dyspnea that was maintained as long as 1 year after treatment termination and a short-term improvement of pulmonary function, CYC may be useful in some SSc-ILD patients. Thus, in SSc-ILD patients experiencing dyspnea and reduced FVC, a short-term use of CYC may be considered in consultation with an expert.
Regarding drugs other than CYC, first, a study showed that steroid therapy (0.5–1 kg/kg of prednisolone) could be expected to slow down pulmonary function decline95, though the effect of steroid in treatment of SSc-ILD is controversial. High dose steroid is not recommended because renal crisis can rapidly develop if the daily dose of prednisolone is over 10 mg86.
MMF has been known to suppress lymphoproliferation and to have an antifibrotic effect. A recently presented randomized comparison group study, Scleroderma Lung Study II (SLS II), compared a patient group administered with MMF (1,500 mg bid) for 24 months and a patient group administered with CYC (2 mg/kg/day) for 12 months. There was little difference in the speed of FVC reduction between the MMF group and the CYC group, and the counts of leucocytes and platelets in the MMF group did not decrease as much as in the CYC group91. Based on the findings, MMF may be considered as initial and maintenance therapies in the treatment of SSc-ILD.
Lately, hematopoietic stem cell transplantation (HSCT) has been attempted in SSc-ILD patients. A randomized controlled trial that compared the effects of HSCT and CYC reported that HSCT helped improve long-term event-free survival and overall survival, but recurrence rate was higher96. Accordingly, HSCT may be considered in cases in which patients did not respond to immunosuppressive therapy or the disease had progressed.
In an open-label randomized controlled trial that investigated rituximab (a B cell depletion therapy) in SSc-ILD, the drug showed an effect of increasing FVC and DLCO 1 year after commencement of therapy97. However, the study included only 12 patients in the sample, which is very small. So far, there is not enough evidence regarding rituximab, and large-scale randomized comparison group research is needed98. As for now, it is believed that patients not responding to the currently available immunosuppressives may be tested on rituximab in a limited manner.
Lung transplantation is the only treatment available to prolong survival of SSc-ILD patients not responding to drug therapy. To improve the prognosis following lung transplantation, suitable patient selection is important. A recent study has proposed the following as lung transplantation exclusion criteria: uncontrolled active inflammatory myopathies, active digital ulcerations, severe SSc associated gastrointestinal disease, cardiac arrhythmia, unstable renal function over the last 3 months, and high risk of SSc associated renal crisis99. However, standardized criteria have not yet been established and further research is warranted.
3) Treatment of RA-ILD
(1) PICO for treatment of patients with RA-ILD: In RA-ILD, do steroids and immunosuppressants (MMF, rituximab) slow the worsening of FVC compared with placebo?
(2) Summary of recommendations for the treatment of patients with RA-ILD: We suggest that, in clinically severe cases, the use of the steroid, MMF, and rituximab may be considered in consultation with an expert (recommendation; expert opinion, level of evidence; low).
If RA-ILD deteriorates despite administration of disease modifying antirheumatic drugs (DMARDs: RA treatment) and tumor necrosis factor inhibitors, conventionally high-dose prednisolone therapy is administered100. But, the evidence for the effectiveness and safety of drug therapy is weak100 and comparison group studies have never been conducted to evaluate drugs in RA-ILD patients.
The authors attempted a systematic literature review and meta-analysis on the effects of steroid and immunosuppressive therapy (MMF and rituximab) in RA-ILD, but could not perform comparative analysis because no study compared steroid or immunosuppressive therapy to placebo. Below are the findings reported in a few single-group studies that used steroid or immunosuppressive therapy such as MMF and rituximab in RA-ILD patients. In a retrospective cohort consisting of 40 RA-ILD patients, baseline FVC improved when prednisolone (1 mg/kg/day) was administered for 6 weeks and then tapered down to 10 mg/day while a DMARD was administered during a follow-up over 6–8 months101. A retrospective single-group study was conducted with 18 CTD-ILD patients including some with RA-ILD and it was found that pulmonary function stabilized and prednisolone maintenance dose decreased during a median of 2.5-year follow-up after treatment with MMF90. A prospective cohort study102 and two retrospective studies103104 with RA-ILD patients reported that rituximab stabilized pulmonary function. Also, it was reported that when rituximab (1,000 mg at day 1, 15, and again at weeks 24 and 26, with methotrexate) was used concomitantly with methotrexate, FVC was maintained in six out of seven RA-ILD patients after 48 weeks102. Also, some case studies reported that azathioprine105, abatacept106, tocilizumab107, and infliximab108 each stabilized pulmonary function in RA-ILD patients.
There is no clear consensus as to when to initiate treatment with the drugs mentioned above. Since no other drug has so far been demonstrated to be effective in RA-ILD, the use of steroids, MMF, or rituximab may be considered for clinically severe cases, in consultation with a specialist.
Go to :
Prognosis of CTD-ILD
The prognosis of CTD-ILD patients varies according to the type of causative CTD. RA-ILD comprises 10%–20% of deaths among RA patients109, and 10% of deaths among all ILD patients789. In SSc-ILD, it is reported that 10-year survival following diagnosis lies in the range 29%–69%. and that in 45%–55% of the patients, pulmonary function deteriorates within the first 3 years, and in 16% severe restrictive pulmonary disease develops1. In Sjögren's syndrome the 5-year survival is reported to be 84%110.
If ILD progresses in spite of immunosuppressive therapy, lung transplantation may be considered. The prognosis of CTD-ILD after lung transplantation is reported to be not very different from that of lung transplantation in IIP patients.
Go to :
 XML Download
XML Download