

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Thrombotic microangiopathies (TMAs) are occlusive micro/macrovascular diseases that often present with intraluminal thrombus formation. TMAs are also clinically described as microangiopathic hemolytic anemia and thrombocytopenia, for which the main forms are thrombotic thrombocytopenic purpura (TTP) and atypical hemolytic uremic syndrome (aHUS) [12]. TTP is a hematological disorder with renal or neurological involvement and platelet-rich thrombi in small vessels [1]. The major pathogenesis of TTP involves deficient ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif, member 13) activity resulting from either a genetic abnormality or the presence of inhibitory antibodies [2]. A deficiency of ADAMTS13 activity results in the accumulation of ultra-large multimers of von Willebrand factor (VWF) due to the loss of cleavage of the 842Tyr-843Met peptide bonds in the A2 domain [3], causing adhesion of the platelets to vascular walls. By contrast, aHUS results from hyperactivity of the alternative complement pathway due to gene mutations producing a loss of regulatory function [i.e. CFH, CFI, CD46 (MCP), or THBD] or a gain of effector function (CFB or C3) [1456]. A loss of regulation that results in unrestricted complement activation leads to endothelial perturbation, leukocyte recruitment, platelet activation, tissue damage, and thrombus formation, eventually leading to end-organ failure [7]. Although the clinical features of TTP and aHUS overlap, neurological manifestations are more prevalent in TTP, whereas aHUS typically has more severe renal involvement. Despite improvements in clinical and research-based approaches, few reliable markers that predict patient outcome have been identified.
This study examined whether complement activation products can be used as biomarkers to differentiate aHUS from TTP and whether they can predict the response to plasma exchange (PEX) treatment and the prognosis, early in the course of therapy. We focused on how complement biomarkers or ADAMTS13 activity are related to the prognosis and clinical outcomes of the patients treated for aHUS.
MATERIALS AND METHODS
Patients
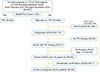
Since 2005, the hematology-oncology laboratory at CHA Bundang Medical Center has been the only nationwide referral center in Korea in charge of ADAMTS13 activity analysis and the collection of data from teaching hospitals for the Korean TTP & TMA registry [8]. Among the 551 patients enrolled in the registry between February 2012 and December 2015, data from those diagnosed with TTP and aHUS were retrospectively analyzed (Fig. 1). Patients with ADAMTS13 activity of <10% (N=48) were selected as the TTP group. Of the remaining 503 patients, 50 Korean patients >18 years of age met the diagnostic criteria of aHUS or were identified by genetic testing (N=9). The diagnoses were confirmed by the central laboratory physician (DO) after reviewing the clinical information and by the referring physicians contacted by telephone or e-mail. No patient was treated with Eculizumab (anti-C5 monoclonal antibody) because it was not approved for the treatment of aHUS in Korea during the study period. Control samples were collected from 40 voluntary healthy donors. The study was in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of CHA Bundang Medical Center; all the patients provided written informed consent.
Evaluation and measurements
Clinical information and blood samples were collected at admission (i.e., the first day TTP or aHUS was suspected and before treatment). The collected samples were sent to a central laboratory (CHA Bundang Medical Center, CHA University) for SDS-agarose gel electrophoresis determination of ADAMTS13 activity via VWF degradation [8]. Samples prospectively collected before administration of any intervention were spun down within 30 min (1,500×g for 20 min at room temperature) to obtain aliquots of platelet-poor plasma that were then frozen at −70℃. Complement activation products in aliquots were measured with ELISA for C3a (generalized pathway), factor Bb (alternative), C4d (classical and lectin), and C5b-9 (terminal) (Quidel, San Diego, CA, USA), as well as C5a (terminal) (BD, Franklin Lakes, NJ, USA). ADAMTS13 inhibitor levels were also determined with ELISA (Sekisui Diagnostics, Stamford, CT, USA).
Definitions of TTP and aHUS
The diagnostic criteria for both conditions included microangiopathic hemolytic anemia characterized by schistocytes in the peripheral blood smear and thrombocytopenia (platelet count <150,000/µL), with TTP and aHUS differentiated by ADAMTS13 activity <10% and >10% (with no evidence of Shiga toxin-producing Escherichia coli or other known secondary TMA) [910], respectively. The median value of ADAMTS13 activity levels among aHUS patients was 77%, and we defined the “high ADAMTS13 activity” as the levels higher than that.
Clinical and hematological outcomes
Standardized report forms given 6 months after the blood tests were used to obtain clinical data regarding the PEX treatments and prognoses. The clinical outcomes were defined according to the Oklahoma TTP-HUS registry [11]. It was defined as an improvement in the number of platelets, regardless of the renal function. A hematological response to PEX treatment was considered a platelet count ≥150,000/µL (measured during or ≤1 week after treatment completion). Recurrent thrombocytopenia requiring re-administration of PEX treatment within 30 days was considered hematological exacerbation, whereas normal platelet counts during this time were considered hematological remission. A subsequent recurrence of TTP or aHUS after hematological remission was considered hematological relapse. Disease-associated mortality was assessed 30 days after the PEX treatment was completed.
aHUS genotyping
Genomic DNA from peripheral blood leukocytes was extracted with a G-DEX II kit (iNtRON Biotechnology, Korea) and used for sequencing and restriction fragment length polymorphism analyses for C3 and CFH mutations (Supplementary Table 1). To validate the results, analyses of each polymorphism were repeated for 30% of the PCRs (randomly selected) followed by DNA sequencing on an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA). The quality control samples exhibited 100% concordance. For aHUS patients in whom polymorphisms were not detected, whole-exome sequencing was performed using an Agilent SureSelect All Exon 50 Mb Kit (Agilent Technologies) and the Illumina HiSeq 2500 platform. Five complement-related (C3, CFB, CFH, CFI, and CD46) and three coagulation-related (DGKE, PLG, and THBD) genes were screened. Rare variants (<5%) were validated by Sanger sequencing. Multiplex ligation-dependent probe amplification was used to detect large gene deletions. The effects of missense variants on protein function and/or splice site were predicted with in silico analyses via Sorting Intolerant from Tolerant, Polymorphism Phenotyping v2, and/or Mutation Taster algorithms.
Statistical analysis
Patient groups were compared with Mann-Whitney U, Fisher's exact, or χ2 tests, where appropriate. Differences in complement activity were evaluated using a Kruskal-Wallis test. Data are reported as medians, ranges, and 25th–75th percentiles. Multivariate logistic regression was performed to assess the difference between hematological outcomes and levels of complement activation markers including laboratory features, with odds ratios (ORs) and 95% confidence intervals (CIs) reported. P-values of <0.05 were considered statistically significant. Analyses were performed using SPSS version 20.0 (SPSS Inc., Chicago, IL, USA).
RESULTS
Patient characteristics
A total of 48 patients with TTP and 50 patients with aHUS [including genetically confirmed (GC)-aHUS] were enrolled in our study. Table 1 summarizes the baseline characteristics of the study population. The age and sex distributions were similar among all the groups. The patients with TTP showed more predominant hematologic manifestations than those with aHUS, such as lower platelet counts (median, 13,000 cells/µL vs. 42,000 cells/µL, respectively; P<0.001) and hemoglobin levels (median, 7.9 g/dL vs. 8.7 g/dL, respectively; P=0.05), and higher PLASMIC scores (median, 6 vs. 5, respectively; P<0.001). In contrast, creatinine levels reflecting acute kidney injury were higher in the patients with aHUS (median, 3.14 mg/dL vs. 0.94 mg/dL; P<0.001). The disease groups were not different with respect to serum lactate dehydrogenase levels or clinical outcomes, such as response, remission, relapse, and disease-associated mortality. The median number of the PEX treatments was 6.5 (range, 0–28) and 5 (range, 0–21) in the TTP and aHUS patients, respectively. The majority [44/48 (91.6%)] of the patients with TTP had inhibitors against ADAMTS13, and they all were newly diagnosed patients. Hemodialysis was performed in 34 of 50 (68.0%) aHUS patients and 9 of 48 (18.8%) TTP patients. aHUS-related heterozygous mutations were identified in 9/50 (18%) patients with aHUS (i.e., CFH, 1; CFB, 1; C3, 4; PLG, 2; THBD, 1) (Supplementary Table 2). Complement biomarkers did not differ between the patients regardless of these identified mutations.
TTP is also associated with complement activation
The levels of each of the complement activation products were compared between TTP and aHUS patients (Fig. 2). Patients with aHUS had significantly higher levels of alternate [C3a (P=0.005), C5a (P=0.001), and factor Bb (P<0.001)] and terminal [C5b-9 (P<0.001)] markers than those of the healthy controls. The levels did not differ between the two disease groups except for factor Bb, which was higher in the aHUS patients than in the TTP patients (median, 1,200 ng/µL vs. 760 ng/µL, respectively; P=0.008). However, C4d levels did not significantly differ among the three groups (P=0.285). The results appeared the same in those of GC-aHUS.
Factors predictive of treatment outcomes according to ADAMTS13 activity
ADAMTS13 activity above normal (≥77%) was detected in 25 (50%) aHUS cases (Table 2). Above-normal ADAMTS13 activity was significantly related to better outcomes, such as improved response (OR, 6.769; 95% CI, 1.605–28.542; P=0.005) and remission (OR, 6.000; 95% CI, 1.693–21.262; P=0.004) rates, and reduced rates of exacerbation (OR, 0.242; 95% CI, 0.064–0.916; P=0.031) and disease-associated mortality (OR, 0.155; 95% CI, 0.029–0.813; P=0.017), but was not related to the relapse rate (OR, 2.087; 95% CI, 0.177–24.615; P>0.05).
Table 3 summarizes the results of the multivariate regression for treatment outcomes according to the levels of complement activation products. In patients with aHUS, a high level of ADAMTS13 activity was beneficial for hematologic outcomes (response: OR, 1.050; 95% CI, 1.009–1.093; P=0.016; exacerbation: OR, 0.965; 95% CI, 0.934–0.997; P=0.034). In addition, serum creatinine (response: OR, 1.433; 95% CI, 1.001–2.051; P=0.049) and platelet count (remission: OR, 1.036; 95% CI, 1.006–1.065; P=0.016) were related to better treatment outcomes. In those with TTP, none of the factors were statistically linked to hematologic outcomes.
DISCUSSION
The results of this study show that complement activation via the alternative rather than the classical or lectin pathway is increased in TTP and aHUS patients. Specifically, patients showed higher levels of generalized (C3a), alternate (factor Bb), and terminal (C5a and C5b-9) markers than healthy controls, whereas the levels of C4d were not different. These findings suggest that the classical or lectin pathway complement activation does not contribute to the pathophysiology of TTP and aHUS or differentiate between the two, unlike ADAMTS13 activity. In addition, abnormal complement activation was not associated with poorer prognoses or adverse outcomes in TTP or aHUS patients.
Complement activation reflects the cooperativity among a variety of plasma proteins to fight invading microorganisms and maintain tissue homeostasis [12]. This activation involves three pathways, namely, classical, lectin, and alternative pathways, and is regulated by soluble factors, such as factors H and I, and thrombomodulin (encoded by THBD), and by membrane-bound regulators, including CD35, CD55, and CD59 as well as membrane cofactor protein (encoded by CD46) [13]. Dysregulated activation is the primary contributor to the pathogenesis of aHUS, and suppression via Eculizumab (anti-C5 monoclonal antibody) treatment is effective and safe [4131415].
Relapse of TTP occurs in 20–30% of patients treated with PEX, and recurrence can be fatal [16]. Therefore, the identification of novel mechanisms contributing to TTP will lead to new potentially life-saving therapeutic modalities. The observed initial complement dysregulation in TTP might exacerbate organ damage or result from microcirculation thrombi causing tissue ischemia. Ruiz-Torres et al. [7] first reported on the complement activation in eight TMA patients. Reti et al. [17] confirmed that levels of C3a and C5b-9 are elevated in patients with TTP compared with those in healthy controls and are significantly decreased by treatment with PEX. Subsequently, Westwood et al. [18] demonstrated that complement activation product levels correlated with the levels of anti-ADAMTS13 IgG in TTP patients and PEX treatment significantly decreased the C3a levels. Recently Wu et al. [19] have found that patients with elevated levels of complement activation products had a poor prognosis and the levels were elevated in both TTP patients and aHUS patients. Recent results suggest that the ultra-large vWF multimer binds to complement and is involved in the pathophysiology of the disease [2021]. This suggests that activation of the classical pathway is not a contributor to TTP or aHUS, which is supported by our findings.
Cataland et al. [9] suggested that complement activation biomarkers may be useful to differentiate TTP from aHUS. However, our data showed that the levels of generalized (C3a) and terminal (C5a and C5b-9) markers of complement activation were similar in both diseases, and although the median level of factor Bb was higher in the group of aHUS patients, there was a substantial overlap in levels between the patients with TTP and those with aHUS. The discrepancies between these studies may be due to the differences in the study subjects or a wide variation in the normal range of the complement activation product levels. The response rates to PEX treatment were also remarkably different (70% in our study vs. 38% reported by Cataland et al.) [9]. Thus, the severity of the disease in the patients in the two studies may also be different. Another possible explanation is that similar levels of complement activation may be induced by sepsis or other nonspecific complications of both TTP and aHUS. However, it may be difficult to distinguish between TTP and aHUS by assessing complement activation early in the disease progression at initial presentation, whereas since the activation is sustained in aHUS patients, it is easier to distinguish the two diseases at later stages. The activation may be transient in TTP, as PEX treatment remedies the ultra-large VWF, thereby reducing complement binding [171822]. Serial measurement during remission or after the initial treatment may be a more rational approach to evaluate the status of complement activation and to differentiate TTP from aHUS.
A severe deficiency of ADAMTS13 activity (<10%) is diagnostic for TTP, and the prolonged decrease in activity is related to poor prognosis and recurrence [16]. Moreover, decreases in ADAMTS13 activity and associated poor prognoses have been reported for other diseases, including during severe sepsis [23], malignant and autoimmune diseases, transplantation of solid organs [24], and malignant hypertension [25]. Although deficient ADAMTS13 activity does not contribute to the development of TMAs other than TTP, it has an important prognostic role. We here show that elevated ADAMTS13 activity predicts favorable clinical outcomes for patients with aHUS treated by PEX. The cause of the low ADAMTS13 activity is unknown, but perhaps, it reflects the complement-mediated binding and clearance of ADAMTS13 by VWF. Therefore, the results of our study suggest that ADAMTS13 activity can be used to determine the appropriate treatment options for aHUS patients as a result of differentiation from TTP patients, in addition to serving as a prognostic indicator to determine whether additional therapies, such as Eculizumab, should be administered.
Because of the high mortality associated with TTP, rapid diagnosis and timely treatment are crucial. Researchers at Massachusetts General Hospital recently developed the PLASMIC score as a diagnostic tool [26]. When comparing patients with high risk (score of 6 or 7) and those with low or intermediate risk (score of ≤5), our model predicted severe ADAMTS13 deficiency with a positive predictive value of 76%, negative predictive value of 75%, sensitivity of 73%, and specificity of 78% (Supplementary Table 3). Thus, the PLASMIC score is not sufficient to distinguish TTP from other TMAs, and ADAMTS13 activity in patients should be measured as soon as possible.
The results presented here are based on a retrospective multicenter analysis, which was limited by the relatively small number of patients. Nevertheless, TTP and aHUS are rare diseases, and obtaining larger population sizes may be challenging. Furthermore, differences in blood collection and processing among centers may result in sampling bias; however, the risk of bias is low, because these institutes have participated in a wide variety of studies with standardized blood samples. Of note, the blood samples used for the analyses in this study were collected before the start of PEX treatment. Further research involving repeated blood collection during and after the treatment is needed. Only 18% of the aHUS patients were found to have genetic mutations in our study, whereas approximately 50% of the patients in the previous studies were found to carry the genetic alterations. It seems likely that the main reason for this gap is due to the ethnic differences because other possible factors, such as Shiga-toxin–producing infection, pregnancy, chemotherapy, sepsis, or other clear causes of TMA were thoroughly excluded in our study. Given that only 21.6% of the genetic mutations were detected in the studies on pediatric aHUS patients in Korea [27], ethnic difference is one possible explanation for this gap. Further studies with a larger cohort of patients need to be undertaken to support this possibility.
In summary, the data presented here indicate that the levels of complement activation biomarkers (except factor Bb) are similar among newly diagnosed TTP and aHUS patients. The level of ADAMTS13 activity may be useful not only for diagnosing TTP but also for predicting the clinical outcomes of the aHUS patients under PEX treatment. To the best of our knowledge, this is the first report linking high normal ADAMTS13 activity with improved outcomes of PEX treatments in aHUS patients. Given that a specific therapy exists for the treatment of aHUS, better diagnostic and prognostic tools for early recognition are necessary to improve patient outcomes These measures may distinguish patients requiring more intensive management, and the results presented here lay the groundwork for future multicenter studies aimed at identifying prognostic markers for aHUS and TMA.




 XML Download
XML Download