

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Acute leukemia (AL), one of the most common malignant tumors in children, is classified as acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML), among which ALL accounts for 60–70% and AML for 30–40% of all AL cases.12 Due to aggravated environmental and air pollution, the incidence of childhood AL has increased in recent years.3 AL poses a serious threat to the lives of patients, and its treatment remains an urgent problem in clinic practice.
microRNAs (miRNAs) are a class of small endogenous noncoding RNAs with 19–25 nucleotides in length that modulate gene expression at the post-transcriptional level via base pairing with 3′-untranslated regions (3′-UTRs) of their target genes.45 miRNAs play an extremely important role in biological processes of multiple tumors, such as cell proliferation, apoptosis, migration, cycle and carcinogenesis.67 To date, numerous studies have reported that abnormal expressions of miRNAs are involved in the pathogenesis and development of AL. For example, Huang, et al.8 have reported that miR-519 promotes AML HL-60 cell proliferation via downregulating the expression of RNA-binding protein human antigen R. miR-34a could enhance cell apoptosis and suppress autophagy by targeting HMGB1 in AML cells.7 miR-146a has been identified to play a dual oncogenic and anticancer role in leukemia,9 because it is overexpressed in childhood ALL and AML,10 but downregulated in adult AML,11 However, the precise roles of miR-146a in AL and its potential molecular mechanism remain to be further investigated.
Leukemic inhibitory factor (LIF), a member of the interleukin (IL)-6 family, is involved in the developmental processes of leukemia,12 Ciliary neurotrophic factor receptor (CNTFR) plays an important role in the pathogenesis of blood diseases by binding LIF.12 The downregulation of CNTFR could facilitate Janus-activated kinases/signal transducer and activator of transcription (JAK/STAT) signaling.13 Ip, et al.12 indicated that miR-708 could exert oncogenic effects in leukemia by regulating CNTFR and activating JAK/STAT pathway. However, whether miR-146a affects childhood AL through CNTFR-mediated JAK/STAT pathway is unknown.
In this research, we investigated the effects of miR-146a on childhood AL and its related molecular mechanisms. Our results suggested that miR-146a could promote the proliferation, migration, and invasion of Jurkat and HL-60 cells, as well as inhibit their apoptosis, by downregulating CNTFR and activating the JAK2/STAT3 pathway. The findings of our study may provide a new theoretical foundation for deeply exploring the treatment of childhood AL.
MATERIALS AND METHODS
Clinical samples
From January 2015 to December 2018, there were 39 newly diagnosed AL pediatric patients (28 ALL and 11 AML) recruited for this study, and the inclusion criteria followed the AL diagnosis criteria in the 2016 edition of the World Health Organization.14 Bone marrow samples were obtained from 39 AL children and 10 non-cancer control children treated in the hematology department of Yulin First Hospital. The non-cancer controls were children with no evidence of malignant hematologic diseases. Monocytes were isolated from 5 mL of heparinized bone marrow by density gradient centrifugation at 500× g for 30 min using Ficoll Paque media (Robbins Scientific, Sunnyvale, CA, USA). Subsequently, the monocytes were washed three times using Hank's balanced salt solution (Thermo, Waltham, MA, USA). The project was approved by the Ethics Committee of Yulin First Hospital (2019-3), and it was performed in accordance with the Declaration of Helsinki. All recruited patients and participants provided signed informed consent.
Cell cultures
The human ALL cell line Jurkat, human AML cell line HL-60, and human embryonic kidney cell line HEK-293T were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in 5% CO2 at 37℃. Jurkat and HL-60 cells were cultured in RPMI-1640 (Gibco, Carlsbad, CA, USA) complemented with 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA, USA) and 1% streptomycin/penicillin (Gibco). HEK-293T cells were grown in Dulbecco Modified Eagle Medium (DMEM, Gibco, Carlsbad, CA, USA) supplemented with 10% FBS and 1% streptomycin/penicillin.
Dual luciferase reporter gene assay
Using the TargetScan website, we found that CNTFR was a target gene of miR-146a. We amplified the 3′-UTR of CNTFR containing the miR-146a binding site and then cloned the 3′-UTR fragment into Psi-CHECK2 reporter vector (Promega, Madison, WI, USA) to construct wild Psi-CHECK2-WT-CNTFR-3′-UTR (CNTFR-wt) and mutant Psi-CHECK2-MUT-CNTFR-3′-UTR (CNTFR-mut). For luciferase assay, miR-146a mimics or miR-146a negative control mimics was co-transfected with reporters plasmids into HEK-293T cells by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Based on the differences in transfection sequences, cells were grouped as follows: mutant-type (MT)+mimics group (transfected with mutant-type sequences and miR-146a mimics), MT+negative control (NC) group (transfected with mutant-type sequences and miR-146a negative control mimics), wild-type (WT)+mimics group (transfected with wild-type sequences and miR-146a mimics), and WT+NC group (transfected with wild-type sequences and miR-146a negative control mimics). The luciferase activity was measured using dual luciferase kits (Promega) after 48 h transfection.
Cell transfection assay
Jurkat and HL-60 cells were added to 6-well plates and incubated at 37℃ for 24 h. When cells reached 80% confluence in the plate well, anti-miR-146a (antisense miR-146a oligonucleotide, Thermo), anti-miR-146a negative control (NC, Thermo), CNTFR-siRNA (QIAGEN, Duesseldorf, Germany), CNTFR-siRNA negative control (QIAGEN), miR-146a mimics (GenePharma, Shanghai, China), miR-146a mimics negative control (GenePharma), and miR-146a inhibitor (GenePharma) were cotransfected into Jurkat and HL-60 cells using Lipofectamine® 2000 Reagent (Invitrogen). The transfected Jurkat and HL-60 cells were randomly assigned to eight groups: Mock group (no treatment), mimics-NC group (transfected with miR-146a mimics negative control), miR-146a mimics group (transfected with miR-146a mimics), miR-146a inhibitor group (transfected with miR-146a inhibitor), anti-miR-NC+siNC group (transfected with anti-miR-146a NC and CNTFR-siRNA NC), anti-miR-146a+siNC group (transfected with anti-miR-146a and CNTFR-siRNA NC), anti-miR-NC+siCNTFR group (transfected with anti-miR-146a NC and CNTFR-siRNA), and anti-miR-146a+siCNTFR group (transfected with anti-miR-146a and CNTFR-siRNA). Finally, all cells were cultured in 37℃ incubator for 48 h.
Cell proliferation assay
Cell proliferation of Jurkat and HL-60 cells was measured by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) colorimetric assay (Sigma, St. Louis, MO, USA). In brief, transfected Jurkat and HL-60 cells were seeded into 96-well plates at a density of 5×103 cells/well. At different time points (0, 24, 48, and 72 h), the culture medium was removed, and 20 µL of MTT (5 mg/mL) was added into each well. After incubation at 37℃ for 4 h, the MTT was removed, and absorbance at 495 nm was measured on a microplate reader (Bio-Rad, Hercules, CA, USA).
Cell apoptosis assay
The apoptosis of Jurkat and HL-60 cells was detected by Annexin V-FITC and propidium iodide apoptosis detection kits (Invitrogen). Briefly, at 48 h after transfection, the Jurkat and HL-60 cells were collected, washed three times with phosphate buffer saline, and re-suspended in 1×binding buffer. Then, Annexin V-FITC and propidium iodide were utilized to stain Jurkat and HL-60 cells for 15 minutes at room temperature. Finally, apoptotic cells were analyzed using a flow cytometer (BD Biosciences, San Jose, CA, USA).
Transwell assay
Transwell assay was conducted using transwell chambers (Corning, New York, NY, USA) pre-coated with Matrigel (BD Biosciences). The transfected Jurkat and HL-60 cells (1×105 cells/well) were collected and inoculated to the upper chamber. Then, 500 µL of RPMI-1640 containing 20% FBS was added into the lower chamber. After incubation for 24 h at 37℃, the non-migratory cells were carefully removed. Then, the migrated cells were fixed with 4% paraformaldehyde for 20 min and stained with 0.5% crystal violet dye (Sigma) for 30 min. The number of migrating cells was counted under an optical microscope at 200× magnification.
Real-time fluorogenic PCR assay
As recommendation of the supplier, total RNA of bone marrow tissue from children with ALL and AML was extracted by using TRIzol (Invitrogen). In addition, total RNA of Jurkat and HL-60 cells was extracted. Then, 500 ng of RNA was reverse-transcribed into cDNA by Revert Aid First Strand cDNA Synthesis Kits (Thermo) and measured using quantitative real-time polymerase chain reaction (qRT-PCR) (Bio-Rad) with SYBR green qPCR Master Mix (Thermo). β-actin was employed as the internal control in the quantitative analysis of CFTFR and LIF expressions and U6 as the internal control in the quantitative analysis of miR-146a expression. Primers used for qRT-PCR analysis were as follows: miR-146a (forward) : 5′-CCGCCGTGAGAACTGAATTCCA-3′, (reverse): 5′-GTGCAGGGTCCGAGGT-3′; U6 (forward): 5′-CTCGCTTCGGCAGCACA-3′, (reverse): 5′-AACGCTTCACGAATTTGCGT-3′; CNTFR (forward): 5′-AAGGACCCAGCCCTCAAGAA-3′, (reverse): 5′-TGCTCTAGAGCAATACAGCAAGTTACC-3′; LIF (forward): 5′-CAGCACCACTGAATCACAGA-3′, (reverse): 5′-AGACACGTAAGGAAAACGCATTA-3′; β-actin (forward): 5′-ACACCTTCTACAATGAGCTG-3′, (reverse): 5′-CTGCTTGCTGATCCACATCT-3′.
Western blot analysis
Total proteins were extracted by lysis buffer. Protein samples (50 µg) were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membrane. The membranes were blocked with 5% skimmed milk, followed by overnight incubation at 4℃ with primary antibody (1:500; JAK2, 1:1000; p-JAK2, 1:500; STAT3, 1:1000; p-STAT3, 1:500; GAPDH, 1:3000, Abcam, Cambridge, MA, USA). Afterwards, peroxidase-labeled secondary antibody (anti-rabbit IgG, 1:5000, Cell Signal, Danvers, MA, USA) was used for incubation for 1 h at room temperature. The protein blots were visualized with an enhanced chemiluminescence kit. Finally, the densities of Western blot bands were analyzed using Quantity One 1-D Analysis Software (Bio-Rad).
Statistical analysis
All statistical analyses were performed using SPSS 22.0 software (IBM Corp., Armonk, NY, USA). The results are presented as means±standard deviations. Differences among multiple groups were analyzed by one-way ANOVA followed by Tukey's post hoc test, and data for two groups were compared using Student's t test. All experiments were repeated three times. p<0.05 was considered to be statistically significant.
RESULTS
Expression of miR-146a upregulated and correlated with poor outcomes in children with AL
The results of qRT-PCR showed that the mRNA expression of miR-146a was significantly increased in ALL and AML children, compared with controls (p<0.05). In addition, miR-146a mRNA expression in the ALL group was higher than that in the AML group (p<0.05) (Fig. 1). These results suggested that miR-146a is highly expressed in children with AL.
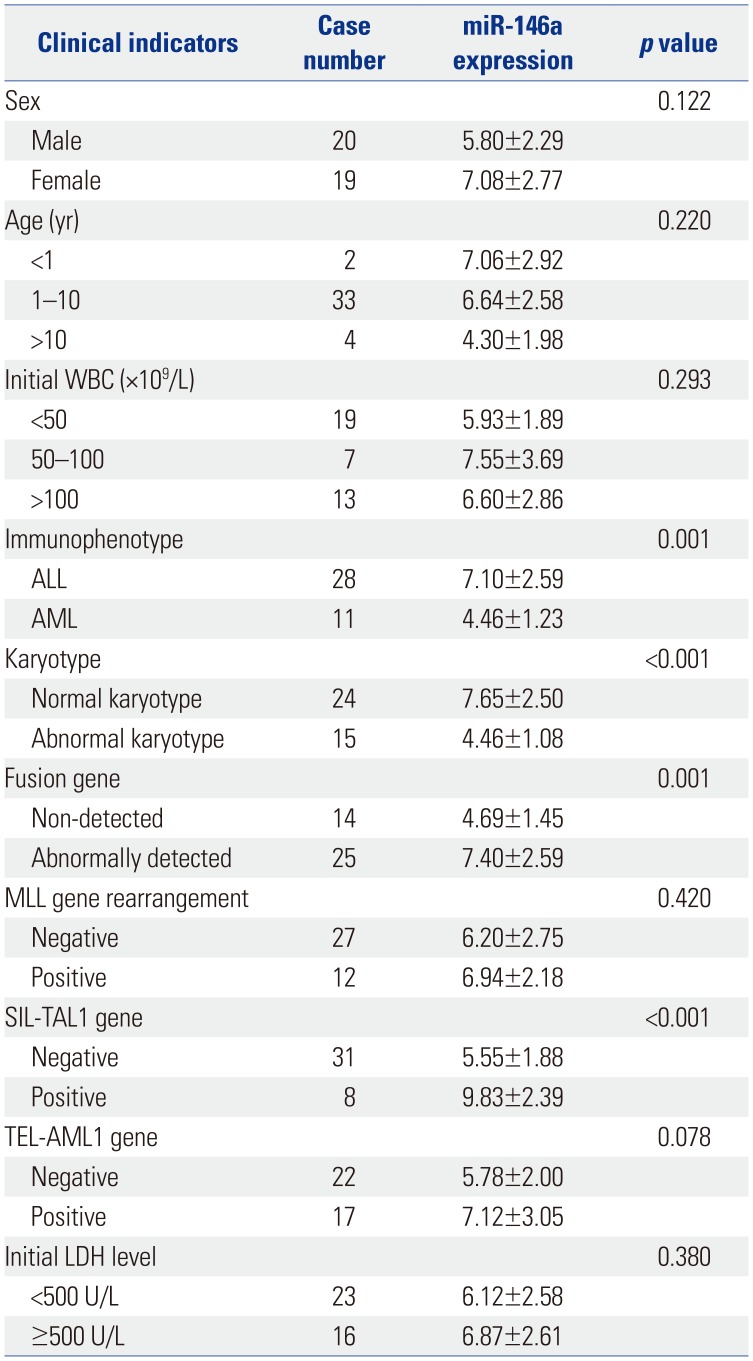
Correlations between the expression of miR-146a and clinical indicators in children with AL are shown in Table 1. The results revealed that the expression of miR-146a is associated with clinical outcomes, such as immunophenotype, karyotype, fusion gene, and SIL-TAL1 gene expression (p<0.05). Other clinic factors, such as age, sex, initial white blood cell counts, MLL gene rearrangement, TEL-AML1, and initial lactate dehydrogenase levels had no significant associations with miR-146a expression (p>0.05).
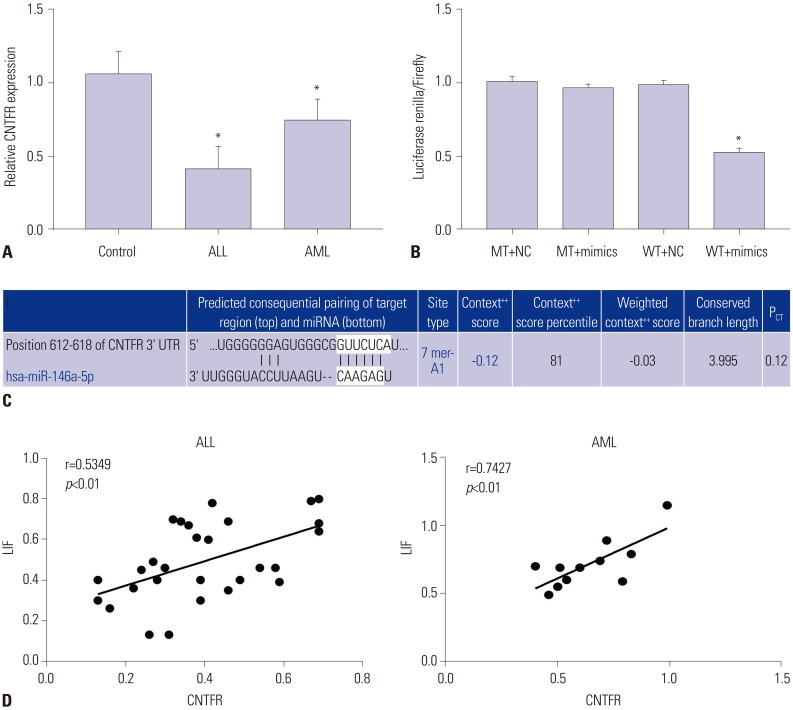
CNTFR a target gene of miR-146a
As shown in Fig. 2A, the mRNA expression levels of CNTFR were remarkedly lower in both ALL and AML groups than in the Control group (p<0.05). In addition, TargetScan predicted that the binding site of miR-146a to CNTFR was the 3′-UTR region (Fig. 2B). According to luciferase reporter assay, we found that luciferase activity in the WT+mimics group was significantly lower than that in the WT+NC group, while the difference in luciferase activity between the MT+NC and MT+mimics groups was not statistically significant (p<0.05) (Fig. 2C). In addition, we also found the expression of LIF was positively correlated with CNTFR expression in ALL and AML children (p<0.01) (Fig. 2D). Thus, we could confirm that miR-146a directly regulates CNTFR expression.
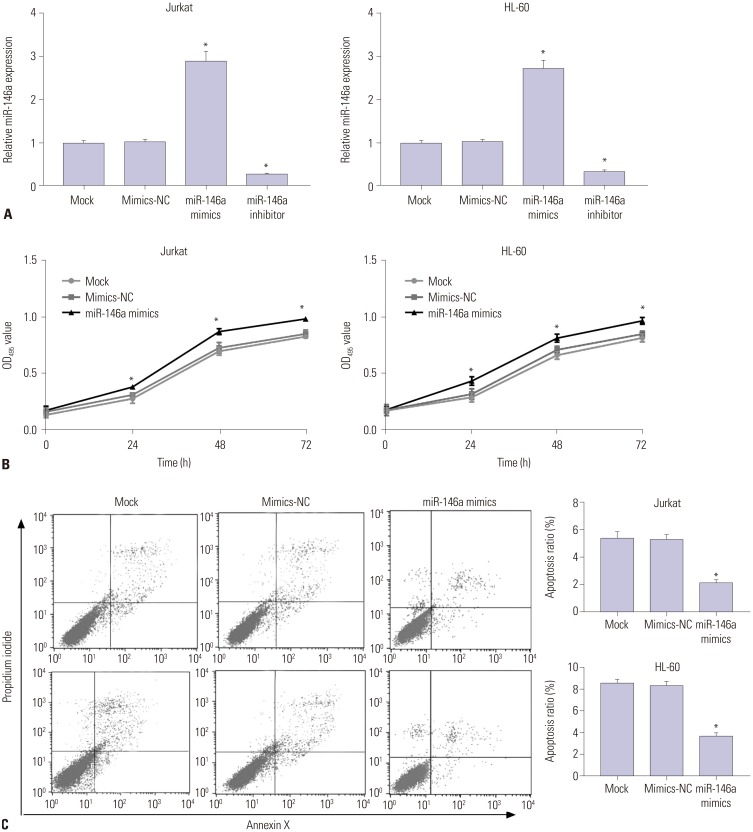
miR-146a promotes cell proliferation and suppresses cell apoptosis of Jurkat and HL-60 cells via downregulating CNTFR
As shown in Fig. 3A, when compared with Mock and the mimics-NC group, the expression of miR-146a in both Jurkat and HL-60 cells was significantly increased in the miR-146a mimics group (p<0.05) and decreased in the miR-146a inhibitor group (p<0.05), suggesting that the transfection was successful. MTT assay showed that the proliferation ability of Jurkat and HL-60 cells in the miR-146a mimics group was markedly higher than that in the Mock and mimics-NC groups (p<0.05) (Fig. 3B). We also found that the apoptosis ability of Jurkat and HL-60 cells in miR-146a mimics group was lower than that in Mock and the mimics-NC group (p<0.05) (Fig. 3C).
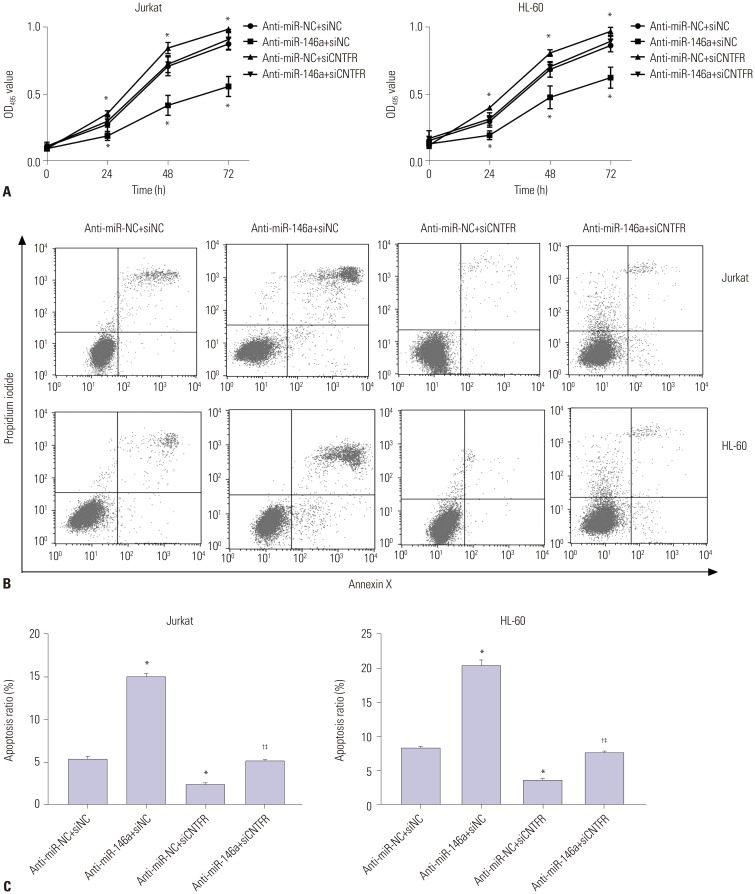
Moreover, Fig. 4A results showed that the cell proliferation ability of Jurkat and HL-60 was significantly decreased in the anti-miR-146a+siNC group, compared with the anti-miR-NC+siNC group (p<0.05). When compared with the anti-miR-NC+siNC group, the cell proliferation abilities of Jurkat and HL-60 cells were markedly increased in the anti-miR-NC+siCNTFR group (p<0.05). However, no significant difference was found between the anti-miR-146a+siCNTFR and anti-miR-NC+siNC groups (p>0.05). All results above indicated that miR-146a could promote cell proliferation of Jurkat and HL-60 cells via downregulating CNTFR.
The results of cell apoptosis assay showed that the cell apoptosis of Jurkat and HL-60 was significantly increased in the anti-miR-146a+siNC group, compared with the anti-miR-NC+siNC group (p<0.05). However, the cell apoptosis of Jurkat and HL-60 was dramatically lower in the anti-miR-NC+siCNTFR group than in the anti-miR-NC+siNC group (p<0.05). When compared with anti-miR-146a+siNC, the cell apoptosis of Jurkat and HL-60 cells was significantly suppressed in the anti-miR-146a+siCNTFR group (p<0.05). In addition, the cell apoptosis of Jurkat and HL-60 cells was markedly higher in the anti-miR-NC+siCNTFR group than in the anti-miR-NC+siCNTFR group (p<0.05) (Fig. 4B and C). All the results suggested that miR-146a could promote cell proliferation and suppress cell apoptosis of Jurkat and HL-60 cells via downregulating CNTFR.
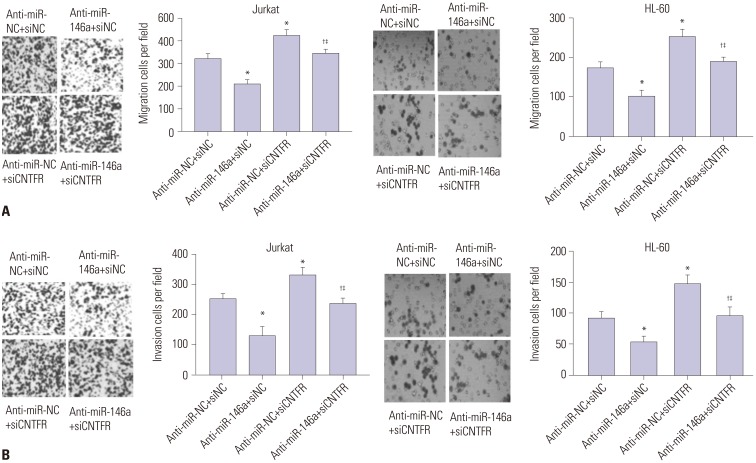
miR-146a increases cell migration and invasion of Jurkat and HL-60 cells by downregulating CNTFR
The impact of miR-146a on cell migration and invasion of Jurkat and HL-60 cells was assessed by transwell assay (Fig. 5). From the results, we observed that counts of migratory and invasive Jurkat and HL-60 cells in the anti-miR-146a+siNC group were both significantly lower than those in the anti-miR-NC+siNC group (p<0.05). In addition, dramatically higher counts of migratory and invasive Jurkat and HL-60 cells in the anti-miR-NC+siCNTFR group were found, compared with those in the anti-miR-NC+siNC group (p<0.05). Compared with anti-miR-146a+siNC, the counts of migratory and invasive Jurkat and HL-60 cells were significantly increased in the anti-miR-146a+siCNTFR group (p<0.05). Moreover, the counts of migratory and invasive Jurkat and HL-60 cells was markedly lower in the anti-miR-NC+siCNTFR group than in the anti-miR-NC+siCNTFR group (p<0.05). All of these suggested miR-146a could stimulate the migration and invasion of Jurkat and HL-60 cells via downregulating CNTFR.
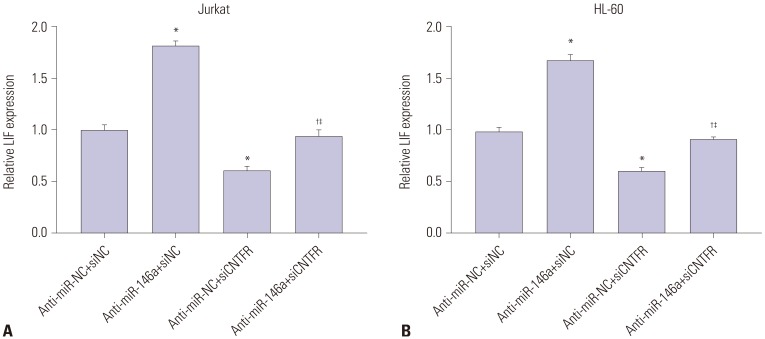
miR-146a inhibits the expression of LIF in Jurkat and HL-60 cells by downregulating CNTFR
The results of qRT-PCR showed that the mRNA expression of LIF in Jurkat and HL-60 cells was significantly greater in anti-miR-146a+siNC group than in the anti-miR-NC+siNC group (p<0.05). On the contrary, LIF mRNA expression in Jurkat and HL-60 cells in the anti-miR-NC+siCNTFR group was markedly lower than that in the anti-miR-NC+siNC group (p<0.05). When compared with anti-miR-146a+siNC, LIF mRNA expression in Jurkat and HL-60 cells was significantly lower than that in the anti-miR-146a+siCNTFR group (p<0.05). Furthermore, mRNA expression of LIF in Jurkat and HL-60 cells was markedly higher in the anti-miR-NC+siCNTFR group than in the anti-miR-NC+siCNTFR group (p<0.05) (Fig. 6), suggesting that miR-146a could inhibit the expression of LIF in Jurkat and HL-60 cells by downregulating CNTFR.
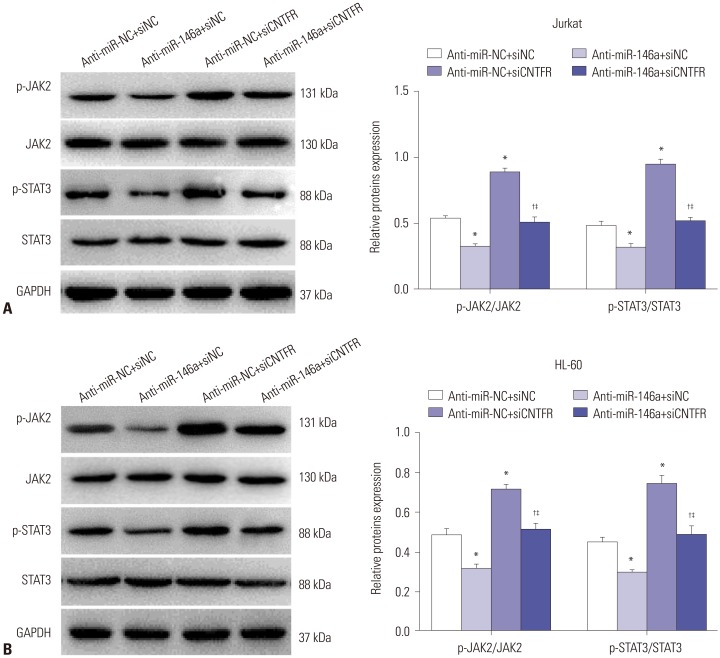
miR-146a activates JAK2/STAT3 signaling by downregulating CNTFR in Jurkat and HL-60 cells
The results of Western blot assay (Fig. 7) showed that the expressions of p-JAK2/JAK2 and p-STAT3/STAT3 in Jurkat and HL-60 cells were significantly decreased in the anti-miR-146a+siNC grouped, compared with the anti-miR-NC+siNC group (p<0.05). However, p-JAK2/JAK2 and p-STAT3/STAT3 expression in Jurkat and HL-60 cells in the anti-miR-NC+siCNTFR group was markedly increased, compared with the anti-miR-NC+siNC group (p<0.05). When compared with anti-miR-146a+siNC, p-JAK2/JAK2 and p-STAT3/STAT3 expression in Jurkat and HL-60 cells was significantly higher in the anti-miR-146a+siCNTFR group (p<0.05). Furthermore, the expressions of p-JAK2/JAK2 and p-STAT3/STAT3 in Jurkat and HL-60 cells were markedly lower in the anti-miR-NC+siCNTFR group than in the anti-miR-NC+siCNTFR group (p<0.05). Altogether, our results revealed that miR-146a may activate the JAK2/STAT3 pathway by downregulating CNTFR in Jurkat and HL-60 cells.
DISCUSSION
AL, a hematopoietic stem cell malignant clonal disease, often occurs in children under 15 years of age.151617 Children with refractory AL have a low survival rate.18 To date, chemotherapy remains the primary treatment method for leukemia; however, a large number of patients experience poor responses to chemotherapy.1920 Thus, it is urgent to explore and discover new molecular approaches in order to better understand this disease and to identify new therapeutic targets. In this study, we demonstrated that miR-146a could promote the proliferation, migration, and invasion and inhibit the apoptosis of Jurkat and HL-60 cells by downregulating CNTFR and activating JAK2/STAT3 signaling.
Accumulating evidence indicates that a variety of miRNAs could play oncogenic or anticancer roles in AL.21 Wang, et al.22 reported that overexpression of miR-125b suppresses AML cell proliferation and invasion and promotes cell apoptosis by targeting the NF-κB pathway. miR-10b has been reported to promote cell proliferation and suppress cell apoptosis in AML by targeting HOXD10. Previous research has confirmed that miR-146a is the only miRNA species that is elevated in both pediatric ALL and AML.23 In our study, miR-146a was highly expressed in children with ALL and AML, which was consistent with previous research. CNTFR is a ganglionic nutrient factor receptor with therapeutic potential in various diseases, such as neurodegenerative diseases,13 cancer,12 and bladder inflammation.24 The downregulation of CNTFR may cause massive AL cell proliferation and apoptosis.12 Our results revealed that CNTFR is lowly expressed in children with ALL and AML. Furthermore, dual luciferase reporter gene assay showed that CNTFR is a target gene of miR-146a. Meanwhile, we also confirmed that miR-146a could promote the proliferation, migration, and invasion and suppress the apoptosis of Jurkat and HL-60 cells by downregulating CNTFR.
Previous studies have revealed that the IL-6 family contains LIF, CNTF, IL-6, IL-11, IL-27, IL-30, IL-31, cardiotrophin-like cytokine, and cardiotrophin-1.2526 Binding of CNTF to its receptor recruits other transmembrane coreceptors (GP130 and LIF) and eventually forms CNTFR-GP130-LIF complexes.27 Ip, et al.12 have indicated that LIF participates in inducing hematopoietic differentiation of leukemia or normal cells. Some researchers have demonstrated that downregulated CNTFR could lead to the loss of LIF hematopoietic differentiation function.12 In this study, we found that LIF expression in Jurkat and HL-60 cells was significantly increased in anti-miR-146a+siNC cells, while the upregulation of LIF could be reversed by siCNTFR, suggesting that miR-146a inhibits the expression of LIF in Jurkat and HL-60 cells by downregulating CNTFR.
The IL-6 type cytokine family (LIF and CNFT) may activate the intracellular signaling cascades, such as JAK/STAT, mitogen-activated protein kinase, and phosphatidylinositol 3′-kinase (PI3K).13 Moreover, JAK/STAT signaling has been reported to be mediated primarily via the IL-6 cytokine family.28 The formation of CNTFR-GP130-LIF complexes is followed by the activation of Janus kinases.29 Subsequently, phosphorylated GP130 and LIFR serve as docking sites for STAT, including STAT3.26 Both aberrant activation of JAK kinase and STAT are associated with a variety of hematological malignancies, such as leukemia.3031 Bromberg, et al.32 also reported that STAT3 is an oncogene that is activated in various cancer cells, including leukemia cells. In addition, miR-147 could regulate the JAK/STAT signaling pathway in myeloid leukemia.33 Some studies have indicated that LIFRα cytoplasmic domain could induce differentiation of the AML cell line HL-60 by inhibiting miR-155 expression through JAK/STAT signaling.34 A study of Ip, et al.12 revealed that miR-708 could exert oncogenic effects in leukemia via regulating CNTFR and activating JAK/STAT pathway. In the present research, miR-146a was found to activate the JAK/STAT pathway by downregulating CNTFR in Jurkat and HL-60 cells, which was consistent with previous research.
In conclusion, our work confirmed that miR-146a is highly expressed and CNTFR is lowly expressed in ALL and AML pediatric patients. In addition, we further demonstrated that miR-146a promotes cell proliferation, migration, and invasion and inhibits cell apoptosis by downregulating CNTFR and activating JAK2/STAT3 pathway in Jurkat and HL-60 cells. Our research highlights a novel regulatory mechanism for miR-146a in Jurkat and HL-60 cells and provides a reference for the treatment of childhood AL. Of course, there are some limitations in this study. In the future, we will investigate the effect of miR-146a on adult leukemia. In addition, we will also assess miR-146a function on the differentiation of leukemia cells.

 XML Download
XML Download